Hi-C Data Normalization and Initial Quality Control, Homer
Ittai Eres
2019-03-12
Last updated: 2020-08-05
Checks: 6 1
Knit directory: HiCiPSC/
This reproducible R Markdown analysis was created with workflowr (version 1.4.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190311) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /Users/ittaieres/HiCiPSC | . |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: code/.DS_Store
Ignored: data/.DS_Store
Ignored: data/TADs/.DS_Store
Ignored: data/TADs/Human_inter_30_KR_contact_domains/.DS_Store
Ignored: data/TADs/scripts/.DS_Store
Ignored: docs/.DS_Store
Ignored: docs/figure/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: Rplot.jpeg
Untracked: Rplot001.jpeg
Untracked: Rplot002.jpeg
Untracked: Rplot003.jpeg
Untracked: Rplot004.jpeg
Untracked: Rplot005.jpeg
Untracked: Rplot006.jpeg
Untracked: Rplot007.jpeg
Untracked: Rplot008.jpeg
Untracked: Rplot009.jpeg
Untracked: Rplot010.jpeg
Untracked: Rplot011.jpeg
Untracked: Rplot012.jpeg
Untracked: Rplot013.jpeg
Untracked: Rplot014.jpeg
Untracked: Rplot015.jpeg
Untracked: Rplot016.jpeg
Untracked: Rplot017.jpeg
Untracked: Rplot018.jpeg
Untracked: Rplot019.jpeg
Untracked: Rplot020.jpeg
Untracked: Rplot021.jpeg
Untracked: Rplot022.jpeg
Untracked: Rplot023.jpeg
Untracked: Rplot024.jpeg
Untracked: Rplot025.jpeg
Untracked: Rplot026.jpeg
Untracked: Rplot027.jpeg
Untracked: Rplot028.jpeg
Untracked: Rplot029.jpeg
Untracked: Rplot030.jpeg
Untracked: Rplot031.jpeg
Untracked: Rplot032.jpeg
Untracked: Rplot033.jpeg
Untracked: Rplot034.jpeg
Untracked: Rplot035.jpeg
Untracked: Rplot036.jpeg
Untracked: Rplot037.jpeg
Untracked: Rplot038.jpeg
Untracked: Rplot039.jpeg
Untracked: Rplot040.jpeg
Untracked: Rplot041.jpeg
Untracked: Rplot042.jpeg
Untracked: Rplot043.jpeg
Untracked: Rplot044.jpeg
Untracked: Rplot045.jpeg
Untracked: Rplot046.jpeg
Untracked: Rplot047.jpeg
Untracked: Rplot048.jpeg
Untracked: Rplot049.jpeg
Untracked: Rplot050.jpeg
Untracked: Rplot051.jpeg
Untracked: Rplot052.jpeg
Untracked: Rplot053.jpeg
Untracked: Rplot054.jpeg
Untracked: Rplot055.jpeg
Untracked: Rplot056.jpeg
Untracked: Rplot057.jpeg
Untracked: Rplot058.jpeg
Untracked: Rplot059.jpeg
Untracked: Rplot060.jpeg
Untracked: Rplot061.jpeg
Untracked: Rplot062.jpeg
Untracked: Rplot063.jpeg
Untracked: Rplot064.jpeg
Untracked: Rplot065.jpeg
Untracked: Rplot066.jpeg
Untracked: Rplot067.jpeg
Untracked: Rplot068.jpeg
Untracked: Rplot069.jpeg
Untracked: Rplot070.jpeg
Untracked: Rplot071.jpeg
Untracked: Rplot072.jpeg
Untracked: Rplot073.jpeg
Untracked: Rplot074.jpeg
Untracked: Rplot075.jpeg
Untracked: Rplot076.jpeg
Untracked: Rplot077.jpeg
Untracked: Rplot078.jpeg
Untracked: Rplot079.jpeg
Untracked: Rplot080.jpeg
Untracked: Rplot081.jpeg
Untracked: Rplot082.jpeg
Untracked: Rplot083.jpeg
Untracked: Rplot084.jpeg
Untracked: Rplot085.jpeg
Untracked: Rplot086.jpeg
Untracked: Rplot087.jpeg
Untracked: Rplot088.jpeg
Untracked: Rplot089.jpeg
Untracked: Rplot090.jpeg
Untracked: Rplot091.jpeg
Untracked: Rplot092.jpeg
Untracked: Rplot093.jpeg
Untracked: Rplot094.jpeg
Untracked: Rplot095.jpeg
Untracked: Rplot096.jpeg
Untracked: Rplot097.jpeg
Untracked: Rplot098.jpeg
Untracked: Rplot099.jpeg
Untracked: Rplot100.jpeg
Untracked: Rplot101.jpeg
Untracked: Rplot102.jpeg
Untracked: Rplot103.jpeg
Untracked: Rplot104.jpeg
Untracked: Rplot105.jpeg
Untracked: Rplot106.jpeg
Untracked: Rplot107.jpeg
Untracked: Rplot108.jpeg
Untracked: Rplot109.jpeg
Untracked: Rplot110.jpeg
Untracked: Rplot111.jpeg
Untracked: Rplot112.jpeg
Untracked: Rplot113.jpeg
Untracked: Rplot114.jpeg
Untracked: Rplot115.jpeg
Untracked: Rplot116.jpeg
Untracked: Rplot117.jpeg
Untracked: Rplot118.jpeg
Untracked: Rplot119.jpeg
Untracked: Rplot120.jpeg
Untracked: Rplot121.jpeg
Untracked: Rplot122.jpeg
Untracked: Rplot123.jpeg
Untracked: Rplot124.jpeg
Untracked: Rplot125.jpeg
Untracked: Rplot126.jpeg
Untracked: Rplot127.jpeg
Untracked: Rplot128.jpeg
Untracked: Rplot129.jpeg
Untracked: Rplot130.jpeg
Untracked: Rplot131.jpeg
Untracked: Rplot132.jpeg
Untracked: Rplot133.jpeg
Untracked: Rplot134.jpeg
Untracked: Rplot135.jpeg
Untracked: Rplot136.jpeg
Untracked: Rplot137.jpeg
Untracked: Rplot138.jpeg
Untracked: Rplot139.jpeg
Untracked: Rplot140.jpeg
Untracked: Rplot141.jpeg
Untracked: Rplot142.jpeg
Untracked: Rplot143.jpeg
Untracked: Rplot144.jpeg
Untracked: Rplot145.jpeg
Untracked: Rplot146.jpeg
Untracked: Rplot147.jpeg
Untracked: Rplot148.jpeg
Untracked: Rplot149.jpeg
Untracked: Rplot150.jpeg
Untracked: Rplot151.jpeg
Untracked: Rplot152.jpeg
Untracked: Rplot153.jpeg
Untracked: Rplot154.jpeg
Untracked: Rplot155.jpeg
Untracked: Rplot156.jpeg
Untracked: Rplot157.jpeg
Untracked: Rplot158.jpeg
Untracked: Rplot159.jpeg
Untracked: Rplot160.jpeg
Untracked: Rplot161.jpeg
Untracked: Rplot162.jpeg
Untracked: Rplot163.jpeg
Untracked: Rplot164.jpeg
Untracked: Rplot165.jpeg
Untracked: Rplot166.jpeg
Untracked: Rplot167.jpeg
Untracked: Rplot168.jpeg
Untracked: Rplot169.jpeg
Untracked: Rplot170.jpeg
Untracked: Rplot171.jpeg
Untracked: Rplot172.jpeg
Untracked: Rplot173.jpeg
Untracked: Rplot174.jpeg
Untracked: Rplot175.jpeg
Untracked: Rplot176.jpeg
Untracked: Rplot177.jpeg
Untracked: Rplot178.jpeg
Untracked: Rplot179.jpeg
Untracked: Rplot180.jpeg
Untracked: Rplot181.jpeg
Untracked: Rplot182.jpeg
Untracked: Rplot183.jpeg
Untracked: Rplot184.jpeg
Untracked: Rplot185.jpeg
Untracked: Rplot186.jpeg
Untracked: Rplot187.jpeg
Untracked: Rplot188.jpeg
Untracked: Rplot189.jpeg
Untracked: Rplot190.jpeg
Untracked: Rplot191.jpeg
Untracked: Rplot192.jpeg
Untracked: Rplot193.jpeg
Untracked: Rplot194.jpeg
Untracked: Rplot195.jpeg
Untracked: Rplot196.jpeg
Untracked: Rplot197.jpeg
Untracked: Rplot198.jpeg
Untracked: Rplot199.jpeg
Untracked: Rplot200.jpeg
Untracked: Rplot201.jpeg
Untracked: Rplot202.jpeg
Untracked: Rplot203.jpeg
Untracked: Rplot204.jpeg
Untracked: Rplot205.jpeg
Untracked: Rplot206.jpeg
Untracked: Rplot207.jpeg
Untracked: Rplot208.jpeg
Untracked: Rplot209.jpeg
Untracked: Rplot210.jpeg
Untracked: Rplot211.jpeg
Untracked: Rplot212.jpeg
Untracked: Rplot213.jpeg
Untracked: Rplot214.jpeg
Untracked: Rplot215.jpeg
Untracked: Rplot216.jpeg
Untracked: Rplot217.jpeg
Untracked: Rplot218.jpeg
Untracked: Rplot219.jpeg
Untracked: Rplot220.jpeg
Untracked: Rplot221.jpeg
Untracked: Rplot222.jpeg
Untracked: Rplot223.jpeg
Untracked: Rplot224.jpeg
Untracked: Rplot225.jpeg
Untracked: Rplot226.jpeg
Untracked: Rplot227.jpeg
Untracked: Rplot228.jpeg
Untracked: Rplot229.jpeg
Untracked: Rplot230.jpeg
Untracked: Rplot231.jpeg
Untracked: Rplot232.jpeg
Untracked: Rplot233.jpeg
Untracked: Rplot234.jpeg
Untracked: Rplot235.jpeg
Untracked: Rplot236.jpeg
Untracked: Rplot237.jpeg
Untracked: Rplot238.jpeg
Untracked: Rplot239.jpeg
Untracked: Rplot240.jpeg
Untracked: Rplot241.jpeg
Untracked: Rplot242.jpeg
Untracked: Rplot243.jpeg
Untracked: Rplot244.jpeg
Untracked: Rplot245.jpeg
Untracked: Rplot246.jpeg
Untracked: Rplot247.jpeg
Untracked: Rplot248.jpeg
Untracked: Rplot249.jpeg
Untracked: Rplot250.jpeg
Untracked: Rplot251.jpeg
Untracked: Rplot252.jpeg
Untracked: Rplot253.jpeg
Untracked: Rplot254.jpeg
Untracked: Rplot255.jpeg
Untracked: Rplot256.jpeg
Untracked: Rplot257.jpeg
Untracked: Rplot258.jpeg
Untracked: Rplot259.jpeg
Untracked: Rplot260.jpeg
Untracked: Rplot261.jpeg
Untracked: Rplot262.jpeg
Untracked: Rplot263.jpeg
Untracked: Rplot264.jpeg
Untracked: Rplot265.jpeg
Untracked: Rplot266.jpeg
Untracked: Rplot267.jpeg
Untracked: Rplot268.jpeg
Untracked: Rplot269.jpeg
Untracked: Rplot270.jpeg
Untracked: Rplot271.jpeg
Untracked: Rplot272.jpeg
Untracked: Rplot273.jpeg
Untracked: Rplot274.jpeg
Untracked: Rplot275.jpeg
Untracked: Rplot276.jpeg
Untracked: Rplot277.jpeg
Untracked: Rplot278.jpeg
Untracked: Rplot279.jpeg
Untracked: Rplot280.jpeg
Untracked: Rplot281.jpeg
Untracked: Rplot282.jpeg
Untracked: Rplot283.jpeg
Untracked: Rplot284.jpeg
Untracked: Rplot285.jpeg
Untracked: Rplot286.jpeg
Untracked: Rplot287.jpeg
Untracked: Rplot288.jpeg
Untracked: Rplot289.jpeg
Untracked: Rplot290.jpeg
Untracked: Rplot291.jpeg
Untracked: Rplot292.jpeg
Untracked: Rplot293.jpeg
Untracked: Rplot294.jpeg
Untracked: Rplot295.jpeg
Untracked: Rplot296.jpeg
Untracked: Rplot297.jpeg
Untracked: Rplot298.jpeg
Untracked: Rplot299.jpeg
Untracked: Rplot300.jpeg
Untracked: Rplot301.jpeg
Untracked: Rplot302.jpeg
Untracked: Rplot303.jpeg
Untracked: Rplot304.jpeg
Untracked: S2A.jpeg
Untracked: S2B.jpeg
Untracked: code/mediation_test.R
Untracked: data/Chimp_orthoexon_extended_info.txt
Untracked: data/Human_orthoexon_extended_info.txt
Untracked: data/Meta_data.txt
Untracked: data/TADs/Arrowhead_individuals/
Untracked: data/TADs/CH.10kb.closest.panTro5
Untracked: data/TADs/CTCF.overlap.computer.sh
Untracked: data/TADs/CTCF/
Untracked: data/TADs/HC.10kb.closest.hg38
Untracked: data/TADs/Human_inter_30_KR_contact_domains_PT6/
Untracked: data/TADs/Rao/
Untracked: data/TADs/TopDom/
Untracked: data/TADs/deprecated/
Untracked: data/TADs/mega.bounds.intersect.c.sh
Untracked: data/TADs/mega.bounds.intersect.merge.sh
Untracked: data/TADs/mega.bounds.rao.sh
Untracked: data/TADs/mega.domains.bedtoolsc.sh
Untracked: data/TADs/mega.domains.rao.sh
Untracked: data/TADs/overlaps/
Untracked: data/TADs/overlaps_rao_style/
Untracked: data/TADs/rao.chimp.subsample.tester.sh
Untracked: data/chimp_lengths.txt
Untracked: data/counts_iPSC.txt
Untracked: data/epigenetic_enrichments/
Untracked: data/final.10kb.homer.df
Untracked: data/final.juicer.10kb.KR
Untracked: data/final.juicer.10kb.VC
Untracked: data/hic_gene_overlap/
Untracked: data/human_lengths.txt
Untracked: data/old_mediation_permutations/
Untracked: output/DC_regions.txt
Untracked: output/IEE.RPKM.RDS
Untracked: output/IEE_voom_object.RDS
Untracked: output/data.4.filtered.lm.QC
Untracked: output/data.4.fixed.init.LM
Untracked: output/data.4.fixed.init.QC
Untracked: output/data.4.init.LM
Untracked: output/data.4.init.QC
Untracked: output/data.4.lm.QC
Untracked: output/full.data.10.init.LM
Untracked: output/full.data.10.init.QC
Untracked: output/full.data.10.lm.QC
Untracked: output/full.data.annotations.RDS
Untracked: output/gene.hic.filt.KR.RDS
Untracked: output/gene.hic.filt.RDS
Untracked: output/gene.hic.filt.VC.RDS
Untracked: output/homer_mediation.rda
Untracked: output/juicer.IEE.RPKM.RDS
Untracked: output/juicer.IEE_voom_object.RDS
Untracked: output/juicer.filt.KR
Untracked: output/juicer.filt.KR.final
Untracked: output/juicer.filt.KR.lm
Untracked: output/juicer.filt.VC
Untracked: output/juicer.filt.VC.final
Untracked: output/juicer.filt.VC.lm
Untracked: output/juicer_mediation.rda
Untracked: output/mc_de.rds
Untracked: output/mc_de_homer.rds
Untracked: output/mc_de_juicer.rds
Untracked: output/mc_node.rds
Untracked: output/mc_node_homer.rds
Untracked: output/mc_node_juicer.rds
Untracked: ~/
Unstaged changes:
Modified: analysis/TADs.Rmd
Deleted: analysis/alt_mediation.Rmd
Modified: analysis/enrichment.Rmd
Modified: analysis/gene_expression.Rmd
Modified: analysis/linear_modeling_QC.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | a34e570 | Ittai Eres | 2020-08-05 | Update formatting for headers |
| html | 2ec8067 | Ittai Eres | 2019-05-09 | Build site. |
| html | 6f6db11 | Ittai Eres | 2019-04-23 | Build site. |
| html | 158b881 | Ittai Eres | 2019-03-13 | Build site. |
| Rmd | 67d6f15 | Ittai Eres | 2019-03-13 | Update directory storage of files; add in data frame of only contacts fixed within species. |
| html | 3d877d7 | Ittai Eres | 2019-03-12 | Build site. |
| Rmd | 607efb8 | Ittai Eres | 2019-03-12 | Initialize website with index and initial QC file. |
First, load necessary libraries: limma, plyr, tidyr, data.table, reshape2, cowplot, plotly, dplyr, Hmisc, gplots, stringr, heatmaply, RColorBrewer, edgeR, tidyverse, and compiler
Initial Data read-in and quality control; Fig S8
First, I read in the data and normalize it with cyclic pairwise loess normalization. These data have come from compiling all of the significant contacts in any individual across species as called by Homer (p<0.01), and then extracting the interaction frequency values for each contact across all individuals. LiftOver was used to convert coordinates between species, with bins being rounded to the nearest 10kb. Here, I look at histograms of the distributions of the contact frequencies on an individual-by-individual basis, to see if they are comparable. I also look at a plot of the mean vs. the variance as an additional quality control metric; this is typically done on RNA-seq count data. In that case, it’s done to check if the normalized data are distributed in a particular fashion (e.g. a poisson model would have a linear mean-variance relationship). In the limma/voom pipeline, count data are log-cpm transformed and a loess trend line of variance vs. mean is then fit to create weights for individual genes to be fed into linear modeling with limma. Since this is not a QC metric typically used for Hi-C data, the only thing I hope to see is no particularly strong relationship between variance and mean.
###Read in data, normalize it with cyclic loess (pairwise), clean it from any rows that have missing values.
setwd("/Users/ittaieres/HiCiPSC")
full.data <- fread("data/final.10kb.homer.df", header=TRUE, stringsAsFactors = FALSE, data.table=FALSE, showProgress = FALSE)
full.contacts <- full.data[complete.cases(full.data[,304:311]), 304:311]
full.contacts <- as.data.frame(normalizeCyclicLoess(full.contacts, span=1/4, iterations=3, method="pairs"))
full.data <- full.data[complete.cases(full.data[,304:311]),]
full.data[,304:311] <- full.contacts
hum.dists <- melt(full.contacts[,c(1:2,5:6)])No id variables; using all as measure variableschi.dists <- melt(full.contacts[,c(3:4,7:8)])No id variables; using all as measure variables#Change names here
hum.dists$variable <- gsub("_norm", "", hum.dists$variable)
chi.dists$variable <- gsub("_norm", "", chi.dists$variable)
###First, a quick look at histograms of the distributions of the significant Hi-C hits from homer, in both humans and chimps.
#FIGS8A
ggplot(data=hum.dists, aes(x=value)) + geom_histogram(binwidth=0.0009, aes(group=variable)) + facet_wrap(~as.factor(variable)) + ggtitle("Distributions of Homer-Significant Hi-C Contacts; Humans") + xlab("Homer-Normalized Interaction Frequency") + coord_cartesian(xlim=c(-6.6, 6.6), ylim=c(0, 4500)) #Human Dist'ns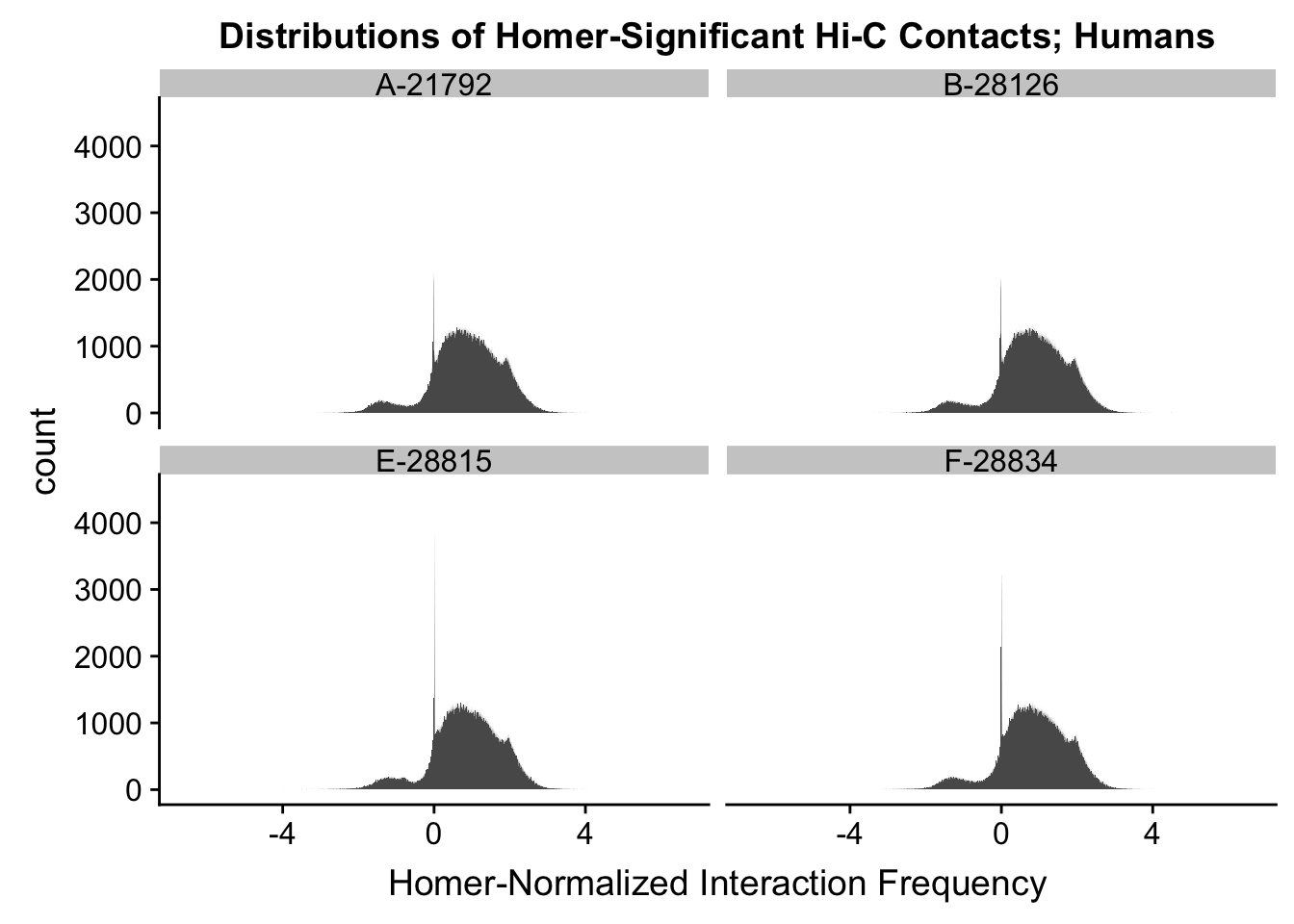
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
FIGS8A <- ggplot(data=hum.dists, aes(x=value)) + geom_histogram(binwidth=0.0009, aes(group=variable)) + facet_wrap(~as.factor(variable)) + ggtitle("Distributions of Homer-Significant Hi-C Contacts; Humans") + xlab("Homer-Normalized Interaction Frequency") + coord_cartesian(xlim=c(-6.6, 6.6), ylim=c(0, 4500)) #Human Dist'ns
#FIGS8B
ggplot(data=chi.dists, aes(x=value)) + geom_histogram(binwidth=0.0009, aes(group=variable)) + facet_wrap(~as.factor(variable)) + ggtitle("Distributions of Homer-Significant Hi-C Contacts; Chimpanzees") + xlab("Homer-Normalized Interaction Frequency") + coord_cartesian(xlim=c(-6.6, 6.6), ylim=c(0, 4500)) #Chimp Dist'ns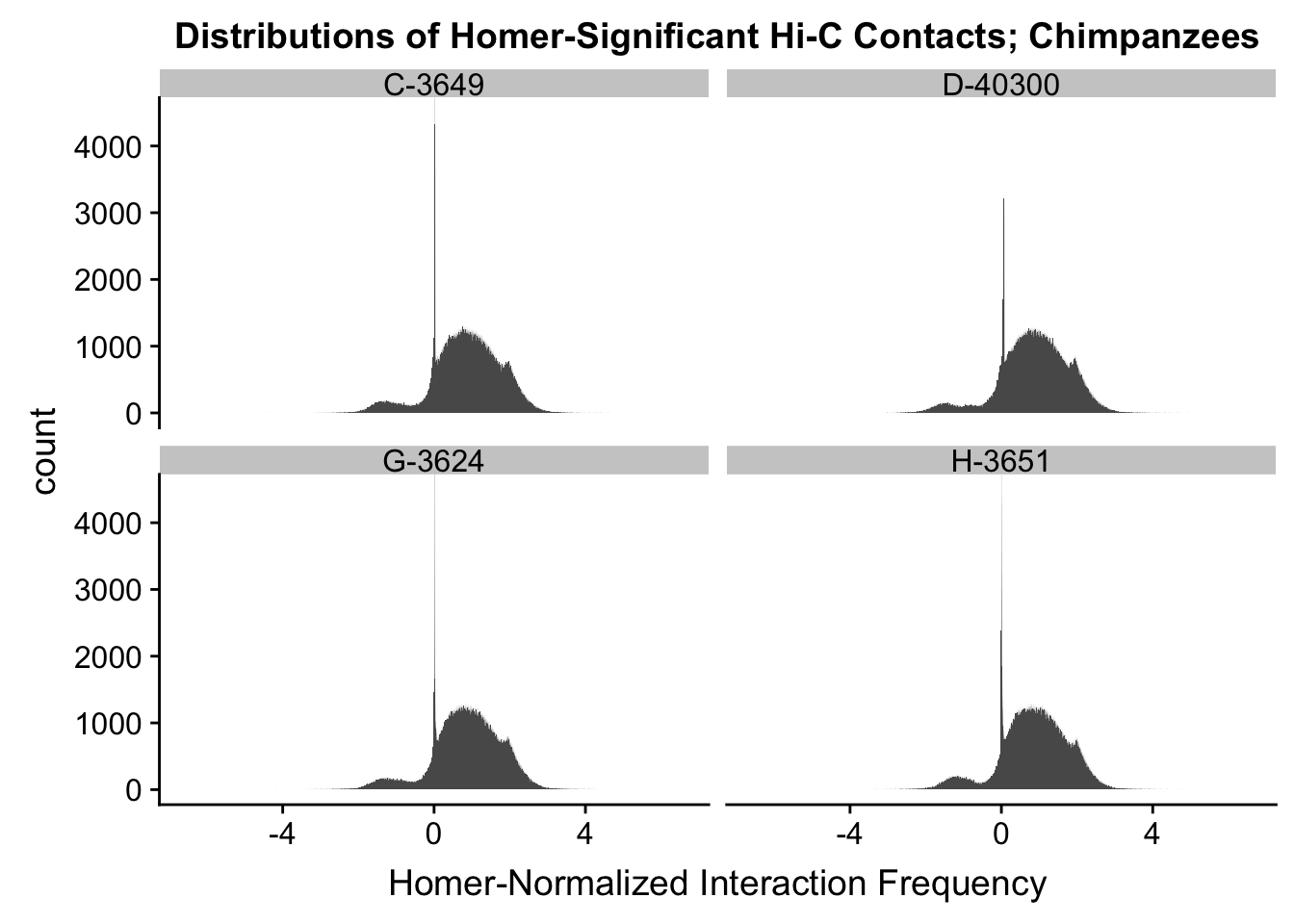
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
FIGS8B <- ggplot(data=chi.dists, aes(x=value)) + geom_histogram(binwidth=0.0009, aes(group=variable)) + facet_wrap(~as.factor(variable)) + ggtitle("Distributions of Homer-Significant Hi-C Contacts; Chimpanzees") + xlab("Homer-Normalized Interaction Frequency") + coord_cartesian(xlim=c(-6.6, 6.6), ylim=c(0, 4500)) #Chimp Dist'ns
#Both sets of distributions look fairly bimodal, with chimps in particular showing a peak around 0.
###As an initial quality control metric, take a look at the mean vs. variance plot for the normalized data. Also go ahead and add on columns for mean frequencies and variances both within and across the species, while they're being calculated here anyway:
select(full.data, "A-21792_norm", "B-28126_norm", "E-28815_norm", "F-28834_norm") %>% apply(., 1, mean) -> full.data$Hmean
select(full.data, "C-3649_norm", "D-40300_norm", "G-3624_norm", "H-3651_norm") %>% apply(., 1, mean) -> full.data$Cmean
select(full.data, "A-21792_norm", "B-28126_norm", "E-28815_norm", "F-28834_norm", "C-3649_norm", "D-40300_norm", "G-3624_norm", "H-3651_norm") %>% apply(., 1, mean) -> full.data$ALLmean #Get means across species.
select(full.data, "A-21792_norm", "B-28126_norm", "E-28815_norm", "F-28834_norm") %>% apply(., 1, var) -> full.data$Hvar #Get human variances.
select(full.data, "C-3649_norm", "D-40300_norm", "G-3624_norm", "H-3651_norm") %>% apply(., 1, var) -> full.data$Cvar #Get chimp variances.
select(full.data, "A-21792_norm", "B-28126_norm", "E-28815_norm", "F-28834_norm", "C-3649_norm", "D-40300_norm", "G-3624_norm", "H-3651_norm") %>% apply(., 1, var) -> full.data$ALLvar #Get variance across species.
{plot(full.data$ALLmean, full.data$ALLvar, main="Mean vs. Variance in Homer-corrected contact frequency", xlab="Mean Hi-C Contact Frequency", ylab="Variance in Hi-C Contact Frequency")
abline(lm(full.data$ALLmean~full.data$ALLvar), col="red")}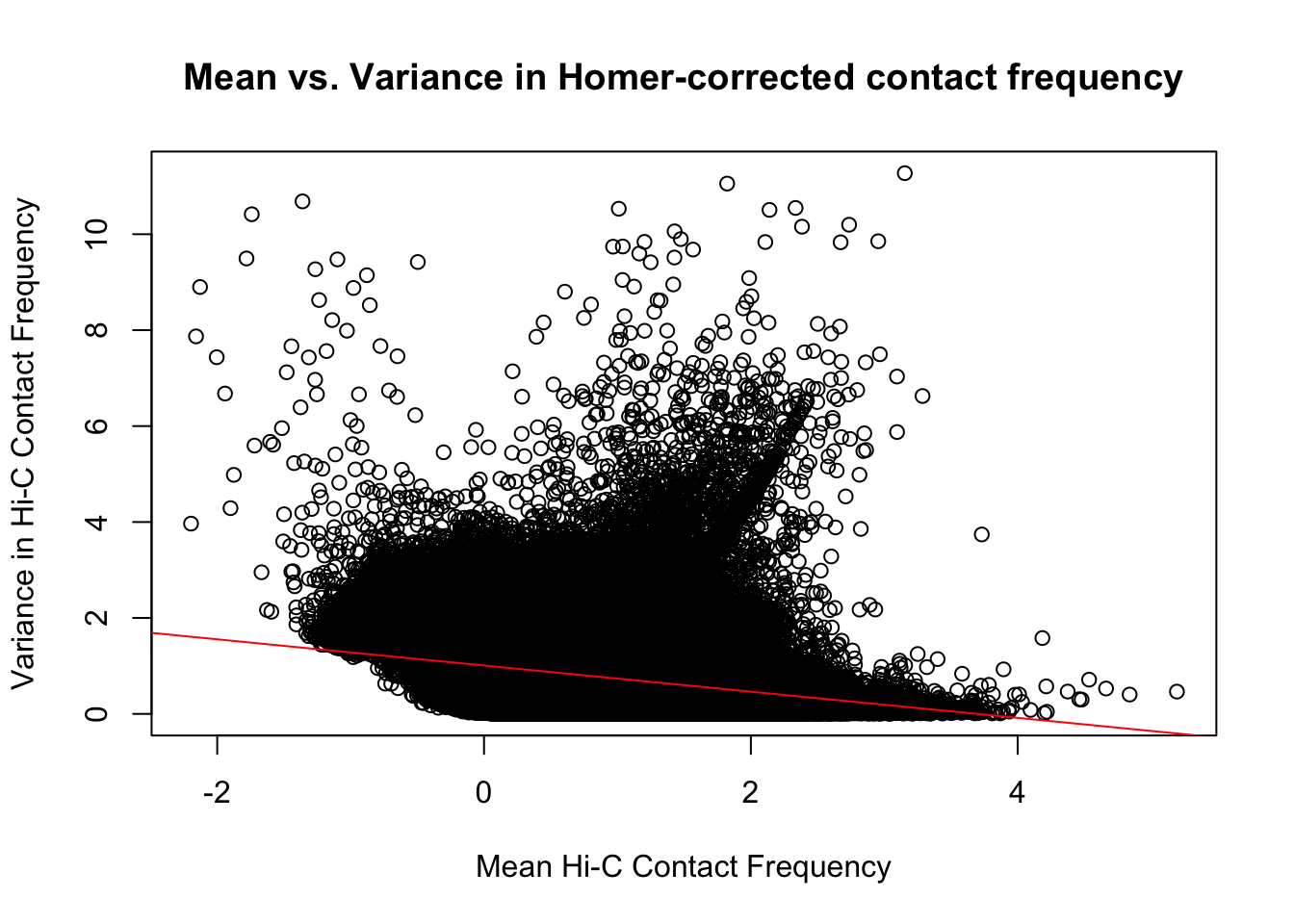
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
#Though there is a slight downward trend, on the whole, there is not a strong correlation between the contact frequency and the variance across individuals for it.
summary(lm(full.data$ALLmean~full.data$ALLvar))$r.squared[1] 0.1076116summary(lm(full.data$ALLmean~full.data$ALLvar))$adj.r.squared[1] 0.1076113On the whole, the distributions look fairly comparable across individuals and species, and the mean vs. variance plots show weak correlation at best between the two metrics. An actual check of the r squared also shows it to be low, at ~0.11. Now knowing the data are comparable in this sense, the next question would be whether they are different enough between the species to separate them from one another with unsupervised clustering and PCA methods.
Data clustering with Principal Components Analysis (PCA) and correlation heatmap clustering.
Now I use PCA, breaking down the interaction frequency values into principal components that best represent the variability within the data. This technique uses orthogonal transformation to convert the many dimensions in variability of this dataset into a lower-dimensional picture that can be used to separate out functional groups in the data. In this case, the expectation is that the strongest principal component, representing the plurality of the variance, will separate out humans and chimps along its axis, grouping the two species independently, as we expect their interaction frequency values to cluster together. I then also compute pairwise pearson correlations between each of the individuals across all Hi-C contacts, and use unsupervised hierarchical clustering to create a heatmap that once again will group individuals based on similarity in interaction frequency values. Again, I would expect this technique to separate out the species very distinctly from one another.
###Now do principal components analysis (PCA) on these data to see how they separate:
meta.data <- data.frame("SP"=c("H", "H", "C", "C", "H", "H", "C", "C"), "SX"=c("F", "M", "M", "F", "M", "F", "M", "F"), "Batch"=c(1, 1, 1, 1, 2, 2, 2, 2), "PE_reads"=c(1084472930, 1103077950, 1015696574, 1047650944, 980287418, 1037054332, 930089380, 964085606), "tags_per_BP"=c(0.351291, 0.357315, 0.342325, 0.353097, 0.317553, 0.335937, 0.313494, 0.324936), "inter_reads"=c(0.1825411, 0.1734168, 0.1559306, 0.2010798, 0.1711258, 0.1479523, 0.1604700, 0.1712287)) #need the metadata first to make this interesting; obtain some of this information from my study design and other parts of it from homer's outputs
pca <- prcomp(t(full.contacts), scale=TRUE, center=TRUE)
#Similar to figure 1A, but done on the whole set of data, without subsetting down to hits found significant in at least 4 individuals (regardless of species).
ggplot(data=as.data.frame(pca$x), aes(x=PC1, y=PC2, shape=as.factor(meta.data$SP), color=as.factor(meta.data$Batch), size=2)) + geom_point() +labs(title="PCA on normalized Hi-C Contact Frequency") + guides(color=guide_legend(order=1), size=FALSE, shape=guide_legend(order=2)) + xlab(paste("PC1 (", signif(100*summary(pca)$importance[2,1],2), "% of variance)")) + ylab((paste("PC2 (", signif(100*summary(pca)$importance[2,2],2), "% of variance)"))) + labs(color="Batch", shape="Species") + scale_shape_manual(labels=c("Chimp", "Human"), values=c(16, 17)) #PCA shows the species separating nicely along PC1, which explains the highest proportion of the variance (as seen from scree plot below). This is nice and suggests that there ARE true differences in the data between species we are likely to detect here. None of the other PCs correlate with the number of PE reads, the tags per base pair statistic, or the fraction of interchromosomal reads, all from the homer output (not shown).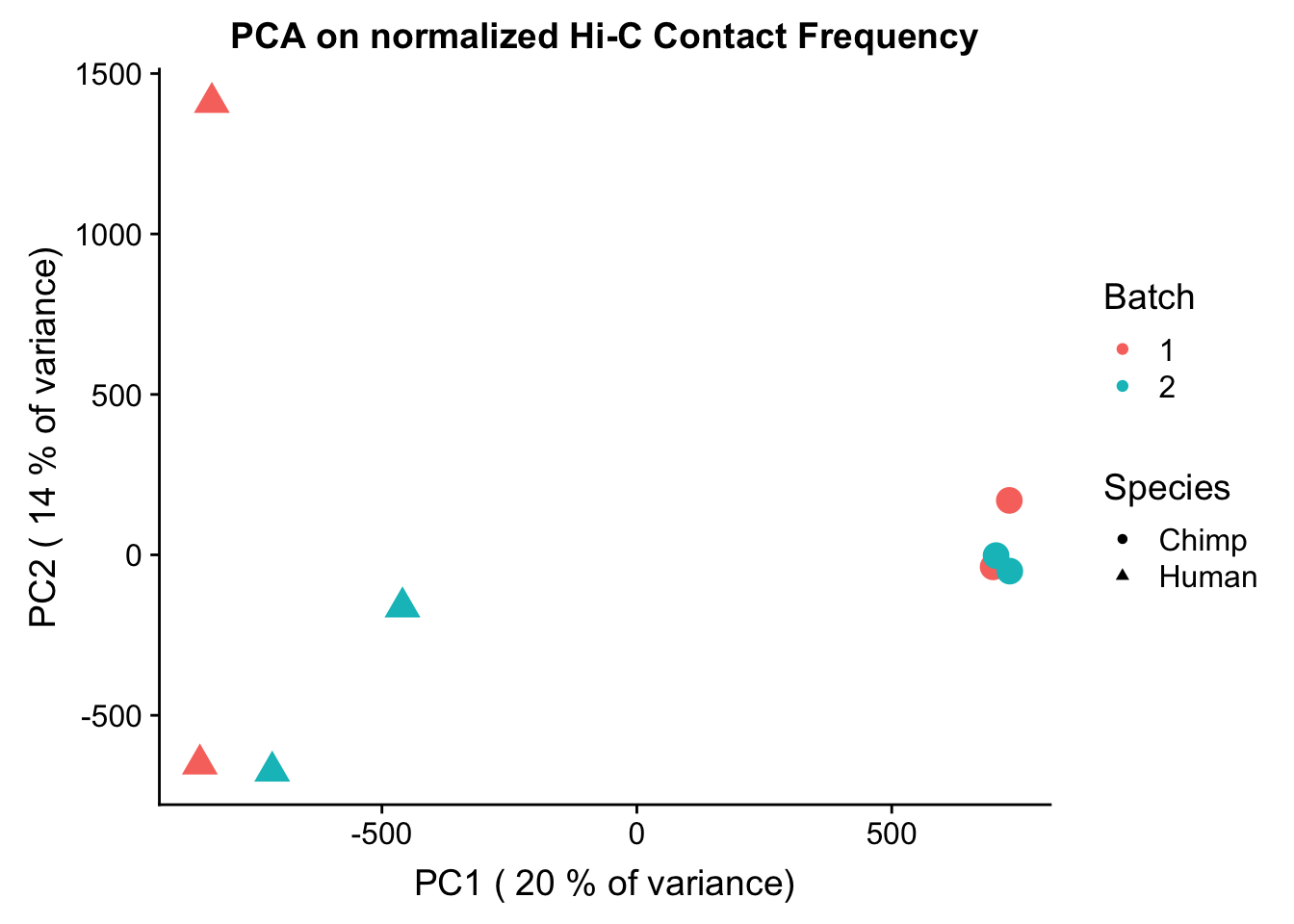
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
barplot(summary(pca)$importance[2,], xlab="PCs", ylab="Proportion of Variance Explained", main="PCA on normalized Hi-C contact frequency") #Scree plot showing all the PCs and the proportion of the variance they explain.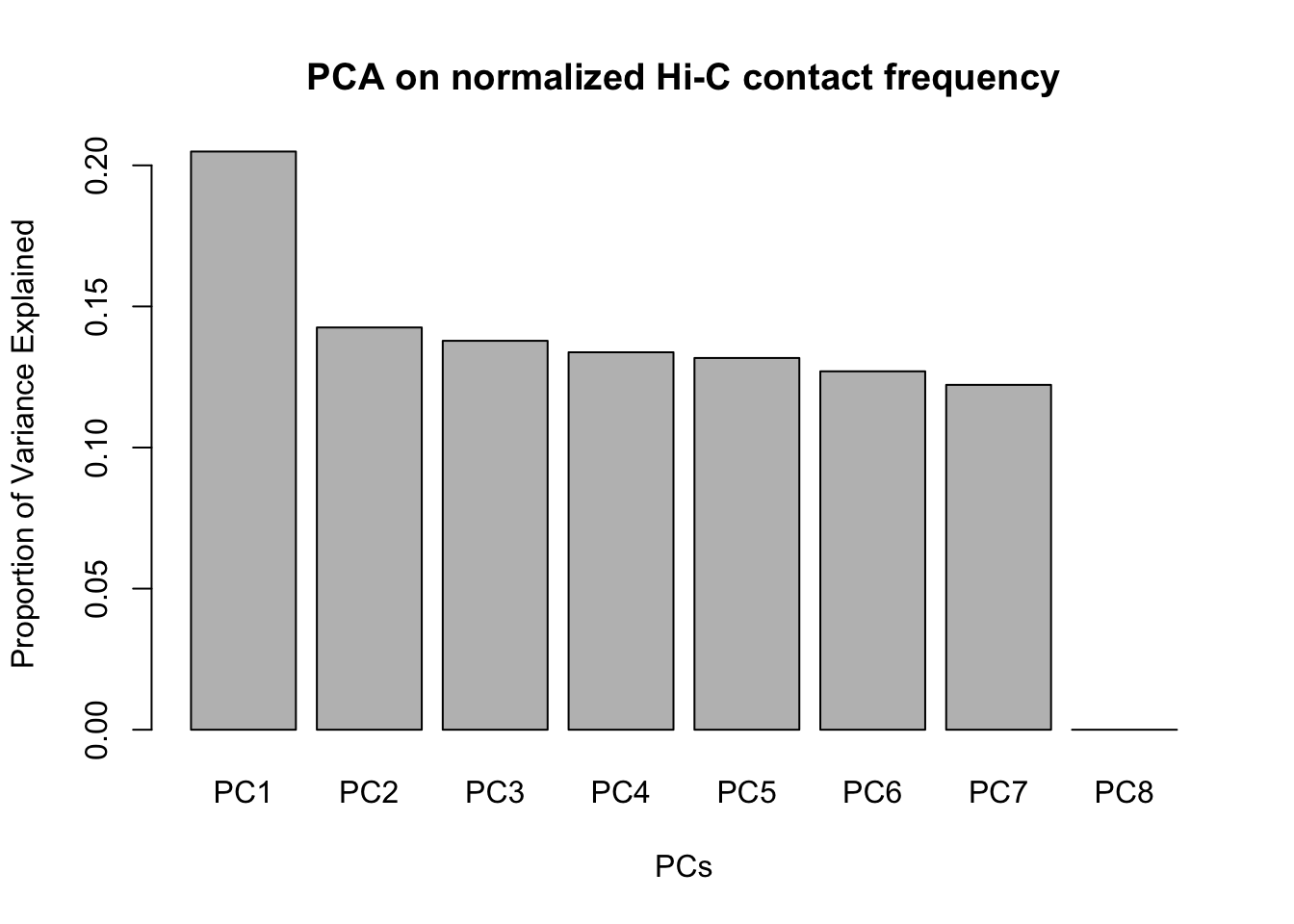
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
###Heatmap clustering of these data as another quality control metric:
corheat <- cor(full.contacts, use="complete.obs", method="pearson") #Corheat for the full data set, and heatmap
#colnames(corheat) <- c("A_HF", "B_HM", "C_CM", "D_CF", "E_HM", "F_HF", "G_CM", "H_CF")
colnames(corheat) <- c("HF1", "HM1", "CM1", "CF1", "HM2", "HF2", "CM2", "CF2") #Better for presentation
rownames(corheat) <- colnames(corheat)
#Similar to figure 1B, but done on the whole set of data, without subsetting down to hits found significant in at least 4 individuals (regardless of species).
heatmaply(corheat, main="Pairwise Pearson Correlation @ 10kb", k_row=2, k_col=2, symm=TRUE, margins=c(50, 50, 30, 30))#We can see that all the humans cluster together, as do all the chimps. This is very good, despite the fact that the correlations are fairly low.Variance exploration; Fig S7
Now I look at several metrics to assess variance in the data, checking which species hits were discovered as significant in and how many individuals a given hit was discovered in. I look at this to understand if there are some cutoffs that should be made to reduce the noisiness of the data and maximize the significance of further findings.
###Add columns to full.data to indicate species of discovery and number of individuals discovered in. These pieces of information are good to know about each of the hits generally, but can also be used to make decisions about filtering out certain classes of hits if variance is associated with any of these metrics.
humNAs <- rowSums(is.na(full.data[,1:149])) #148 is when there is no human info. 37 NAs per individual.
chimpNAs <- rowSums(is.na(full.data[,150:297])) #Same as above of course.
full.data$found_in_H <- (4-humNAs/37) #Set a new column identifying number of humans hit was found in
full.data$found_in_C <- (4-chimpNAs/37) #Set a new column identifying number of chimps hit was found in
full.data$disc_species <- ifelse(full.data$found_in_C>0&full.data$found_in_H>0, "B", ifelse(full.data$found_in_C==0, "H", "C")) #Set a column identifying which species (H, C, or Both) the hit in question was identified in. Works with nested ifelses.
full.data$tot_indiv_IDd <- full.data$found_in_C+full.data$found_in_H #Add a column specifying total number of individuals homer found the significant hit in.
###Take a look at what proportion of the significant hits were discovered in either of the species (or both of them).
sum(full.data$disc_species=="H") #~1.2 million[1] 1186069sum(full.data$disc_species=="C") #~1.1 million[1] 1057817sum(full.data$disc_species=="B") #~690 k[1] 688624#This is reassuring, as similar numbers of discoveries in both species suggests comparable power for detection.
###Now I look at variance in interaction frequency as a function of the number of individuals in which a pair was independently called as significant. Essentially, I'm checking here to see if there is some kind of cutoff I should make for the significant hits--i.e., if the variance looks to drop off significantly after a hit is discovered in at least 2 individuals, this suggests possibly filtering out any hit only discovered in 1 individual.
myplot <- data.frame(disc_species=full.data$disc_species, found_in_H=full.data$found_in_H, found_in_C=full.data$found_in_C, tot_indiv_IDd=full.data$tot_indiv_IDd, Hvar=full.data$Hvar, Cvar=full.data$Cvar, ALLvar=full.data$ALLvar)
ggplot(data=myplot, aes(group=tot_indiv_IDd, x=tot_indiv_IDd, y=ALLvar)) + geom_boxplot() + ggtitle("Number of Individuals a Hi-C Hit is Discovered in Relative to Variance") + xlab("Number of Individuals Where Hi-C Contact Called Significant") + ylab("Variance in Interaction Frequency")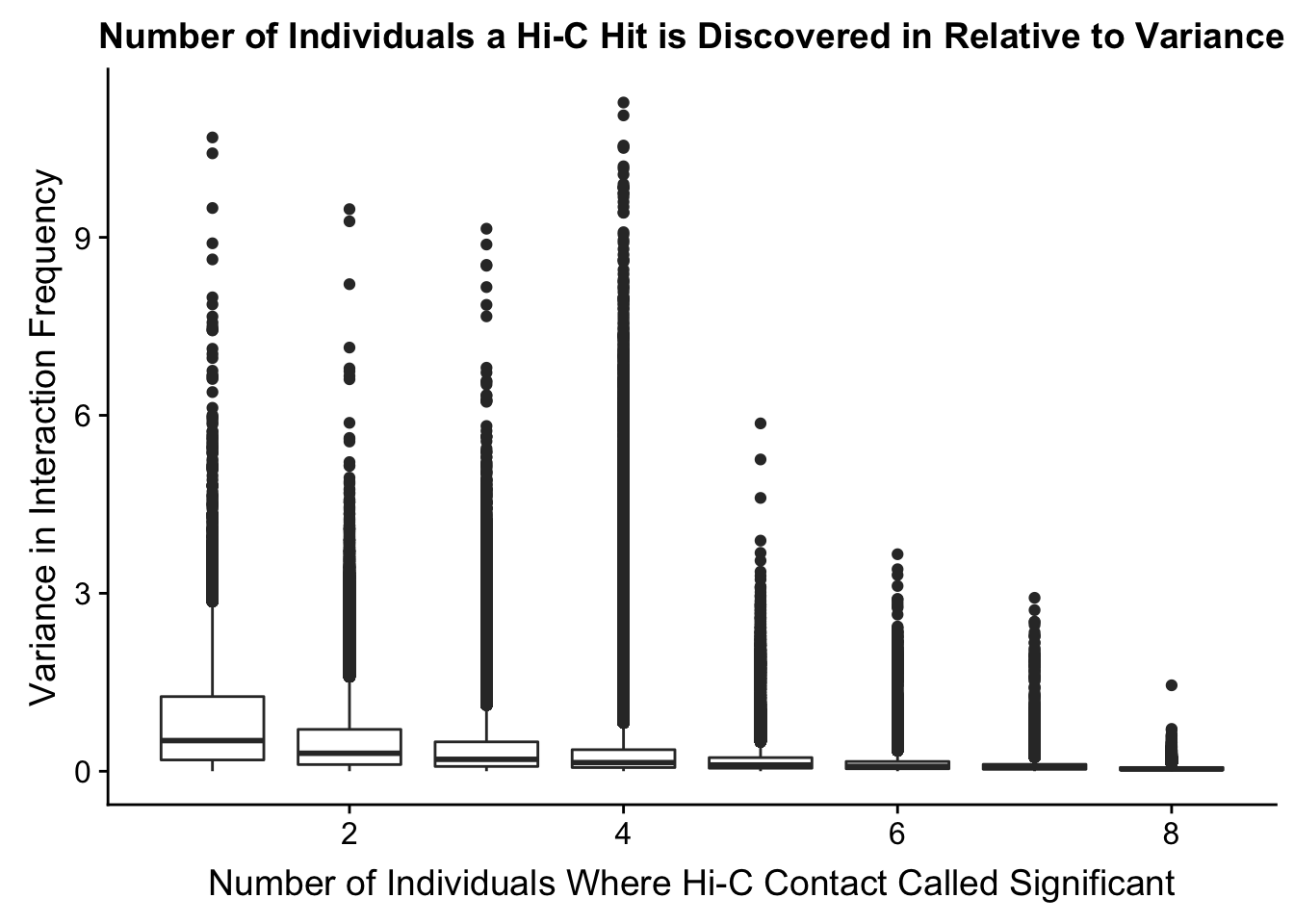
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
#FIGS7
FIGS7A <- ggplot(data=myplot, aes(group=tot_indiv_IDd, x=tot_indiv_IDd, y=ALLvar)) + geom_boxplot() + ggtitle("Number of Individuals a Hi-C Hit is Discovered in Relative to Variance") + xlab("Number of Individuals Where Hi-C Contact Called Significant") + ylab("Variance in Interaction Frequency")
ggplot(data=myplot, aes(group=tot_indiv_IDd, x=tot_indiv_IDd, y=ALLvar)) + geom_boxplot() + scale_y_continuous(limits=c(0, 1)) + ggtitle("Number of Individuals a Hi-C Hit is Discovered in Relative to Variance") + xlab("Number of Individuals Where Hi-C Contact Called Significant") + ylab("Variance in Interaction Frequency")Warning: Removed 737679 rows containing non-finite values (stat_boxplot).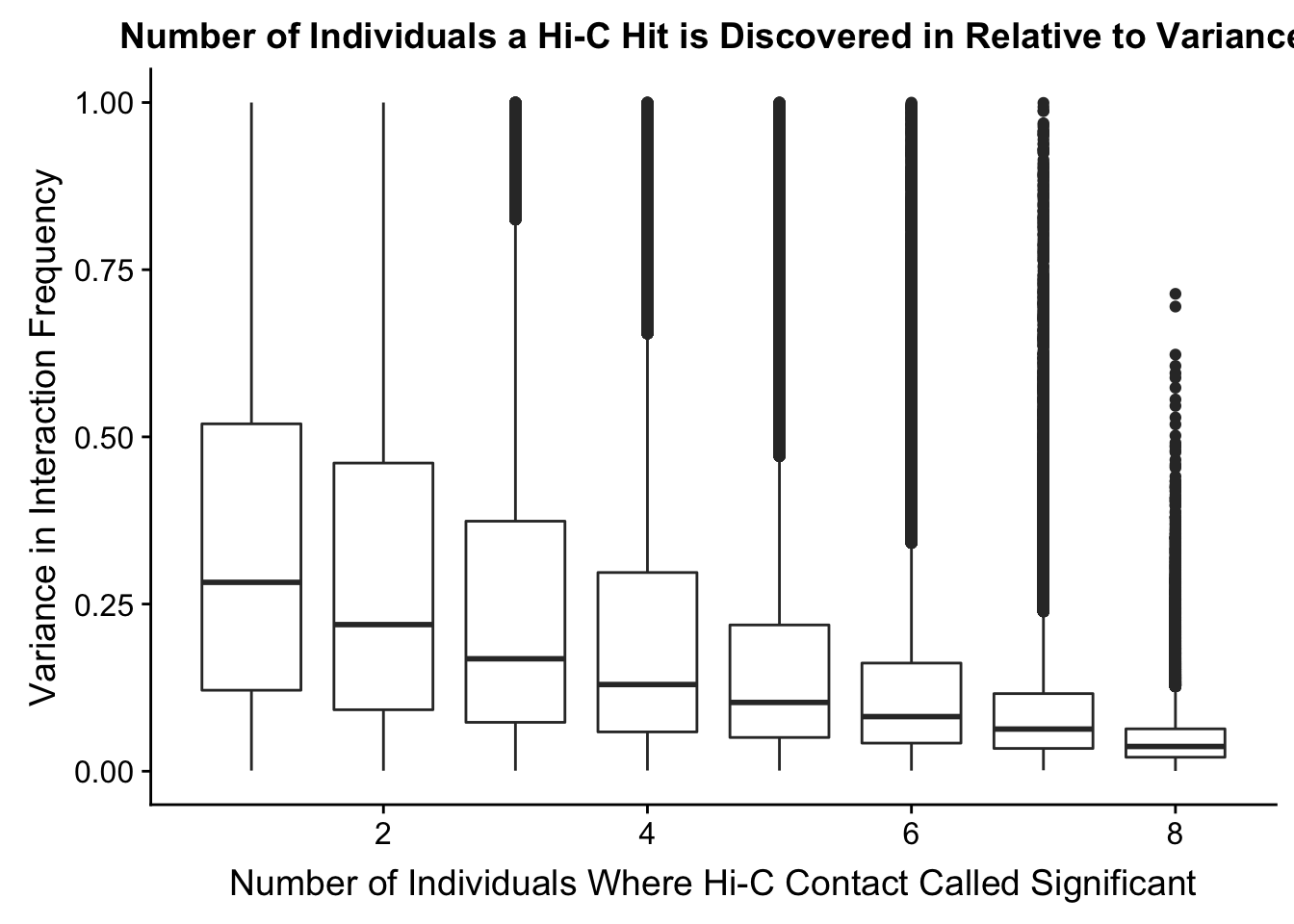
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
FIGS7B <- ggplot(data=myplot, aes(group=tot_indiv_IDd, x=tot_indiv_IDd, y=ALLvar)) + geom_boxplot() + scale_y_continuous(limits=c(0, 1)) + ggtitle("Number of Individuals a Hi-C Hit is Discovered in Relative to Variance") + xlab("Number of Individuals Where Hi-C Contact Called Significant") + ylab("Variance in Interaction Frequency")
FIGS7 <- plot_grid(FIGS7A, FIGS7B, labels=c("A", "B"), align = "h")Warning: Removed 737679 rows containing non-finite values (stat_boxplot).save_plot("~/Desktop/FIGS7A.eps", FIGS7)From this, it would appear that variance drops off drastically after discovery in at least 4 individuals. I observed a similar pattern when looking at number of individuals a hit is discovered in vs. species-specific variances, as well as similar reductions when looking at variance in hits within species vs. number of individuals discovered in (not shown). Hence I now repeat all of the QC analyses done above, but this time conditioned upon discovery in 4 individuals. Moving forward I will ultimately filter out all the hits that were not found in at least 4 individuals, though I sometimes check the full dataset as well just for sanity’s sake.
Initial Quality Control Analyses Conditioned Upon Discovery in at Least 4 Individuals; Fig 1
Given the results just seen for variance, I now repeat all the analyses I carried out previously on the data conditioned upon homer discovering a hit as significant in at least 4 individuals. Based upon a reviewer concern, I also check these things for hits that were discovered as significant in all 4 individuals within a given species (i.e. significant contacts that are not “polymorphic”)
#Repeating all QC with the condition of discovery in 4 individuals:
data.4 <- filter(full.data, tot_indiv_IDd>=4) #Enforcing this condition brings us down from ~3 million hits to ~347k hits.
data.4.fixed <- filter(full.data, found_in_H==4|found_in_C==4) #Enforcing this reviewer-suggested condition of only including contacts significant in all individuals from a species brings us down to ~164k hits.
###See how distributions look with condition of 4 individuals.
ggplot(data=melt(data.4[,c(304:305, 308:309)]), aes(x=value)) + geom_histogram(binwidth=0.0009, aes(group=variable)) + facet_wrap(~as.factor(variable)) + ggtitle("Distributions of Homer-Significant Hi-C Contacts, Humans | 4") + xlab("Homer-Normalized Interaction Frequency") + coord_cartesian(xlim=c(-6.6, 6.6), ylim=c(0, 450))#Human Dist'nsNo id variables; using all as measure variables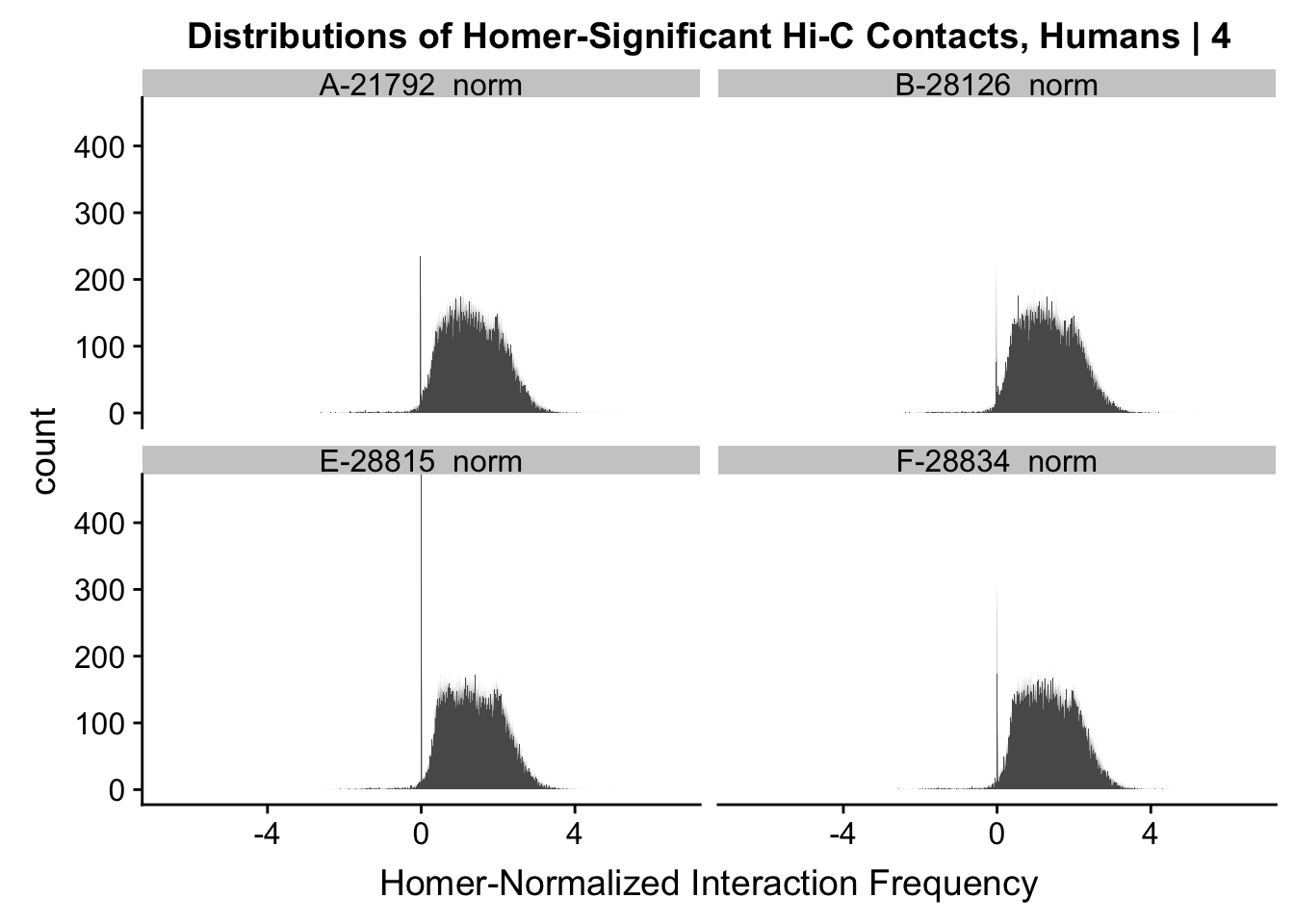
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
ggplot(data=melt(data.4[,c(306:307, 310:311)]), aes(x=value)) + geom_histogram(binwidth=0.0009, aes(group=variable)) + facet_wrap(~as.factor(variable)) + ggtitle("Distributions of Homer-Significant Hi-C Contacts, Chimps | 4") + xlab("Homer-Normalized Interaction Frequency") + coord_cartesian(xlim=c(-6.6, 6.6), ylim=c(0, 450)) #Chimp Dist'nsNo id variables; using all as measure variables
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
#Overall the distributions look fairly similar to before, with the bimodality seen before being much less stark here. Most of the negative interaction frequency significant hits have dropped out, and there is still a strong mass around 0 (especially in the chimp distributions). The majority of the mass of the hits now seems to hover around 1.8 or so.
#Check the same thing but for contacts fixed within species:
ggplot(data=melt(data.4.fixed[,c(304:305, 308:309)]), aes(x=value)) + geom_histogram(binwidth=0.0009, aes(group=variable)) + facet_wrap(~as.factor(variable)) + ggtitle("Distributions of Homer-Significant Hi-C Contacts, Humans | 4 Fixed") + xlab("Homer-Normalized Interaction Frequency") + coord_cartesian(xlim=c(-6.6, 6.6), ylim=c(0, 450))#Human Dist'nsNo id variables; using all as measure variables
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
ggplot(data=melt(data.4.fixed[,c(306:307, 310:311)]), aes(x=value)) + geom_histogram(binwidth=0.0009, aes(group=variable)) + facet_wrap(~as.factor(variable)) + ggtitle("Distributions of Homer-Significant Hi-C Contacts, Chimps | 4 Fixed") + xlab("Homer-Normalized Interaction Frequency") + coord_cartesian(xlim=c(-6.6, 6.6), ylim=c(0, 450)) #Chimp Dist'nsNo id variables; using all as measure variables
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
###Mean vs. variance plot with condition of 4 individuals:
{plot(data.4$ALLmean, data.4$ALLvar, main="Mean vs. Variance in Homer-corrected contact frequency | 4", xlab = "Mean Hi-C Contact Frequency", ylab="Variance in Hi-C Contact Frequency")
abline(lm(data.4$ALLmean~data.4$ALLvar), col="red")}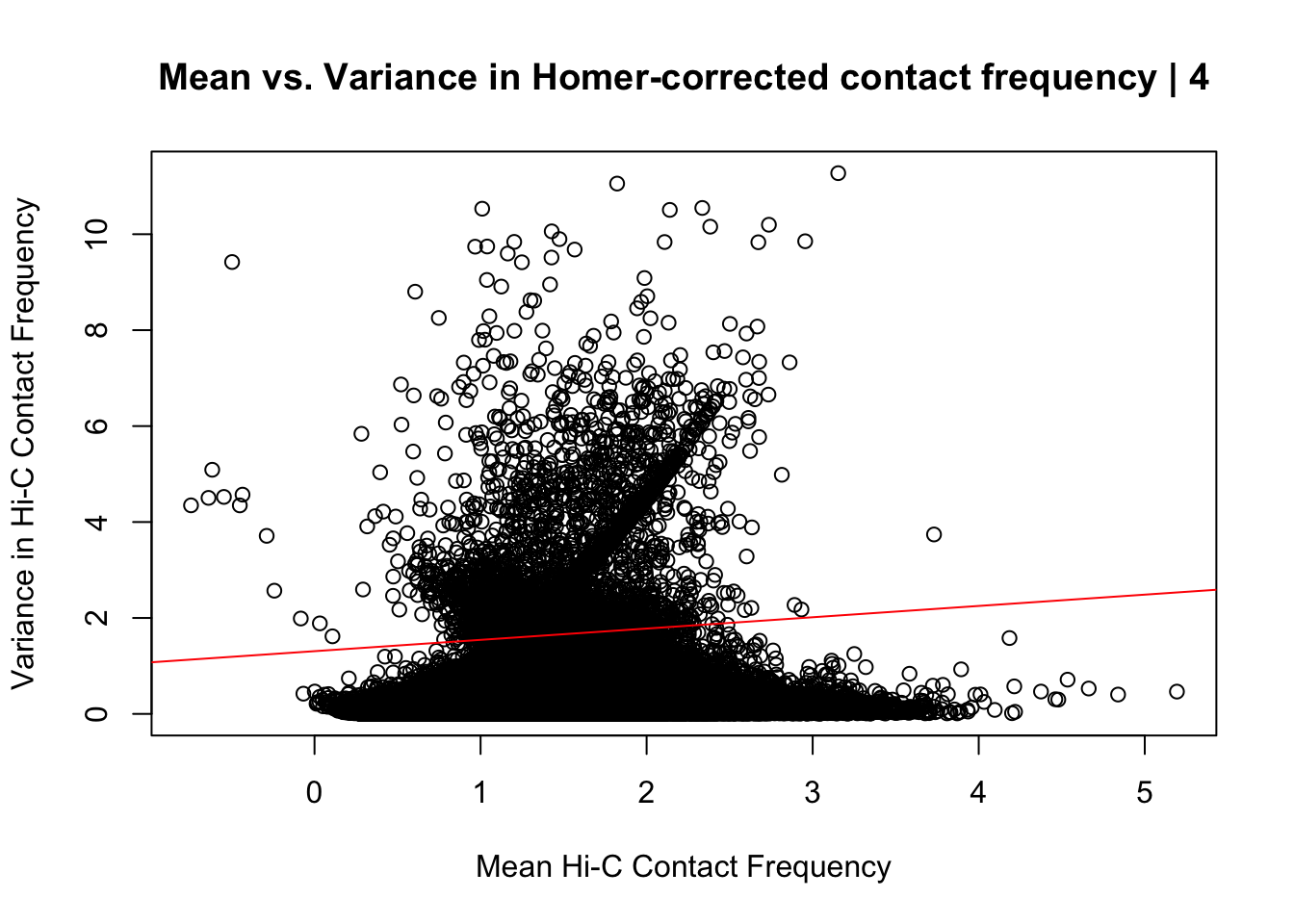
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
summary(lm(data.4$ALLmean~data.4$ALLvar))$r.squared[1] 0.02812765summary(lm(data.4$ALLmean~data.4$ALLvar))$adj.r.squared[1] 0.02812485#Looks very similar to the same plot above for the whole dataset; with slightly fewer outliers in the top left of the plot and a different trend in the red line modeling the mean regressed upon the variance. Here, the line is fairly flat, perhaps trending slightly upwards, whereas the entire dataset sees it have a moderate negative slope. Overall, would really prefer there to be no relationship here, but, again, this is not an ideal metric for Hi-C data, so it's not a particularly worrisome result. the r squared also is quite low (0.028).
#Same for fixed in each species:
{plot(data.4.fixed$ALLmean, data.4.fixed$ALLvar, main="Mean vs. Variance in Homer-corrected contact frequency | 4 Fixed", xlab = "Mean Hi-C Contact Frequency", ylab="Variance in Hi-C Contact Frequency")
abline(lm(data.4.fixed$ALLmean~data.4.fixed$ALLvar), col="red")}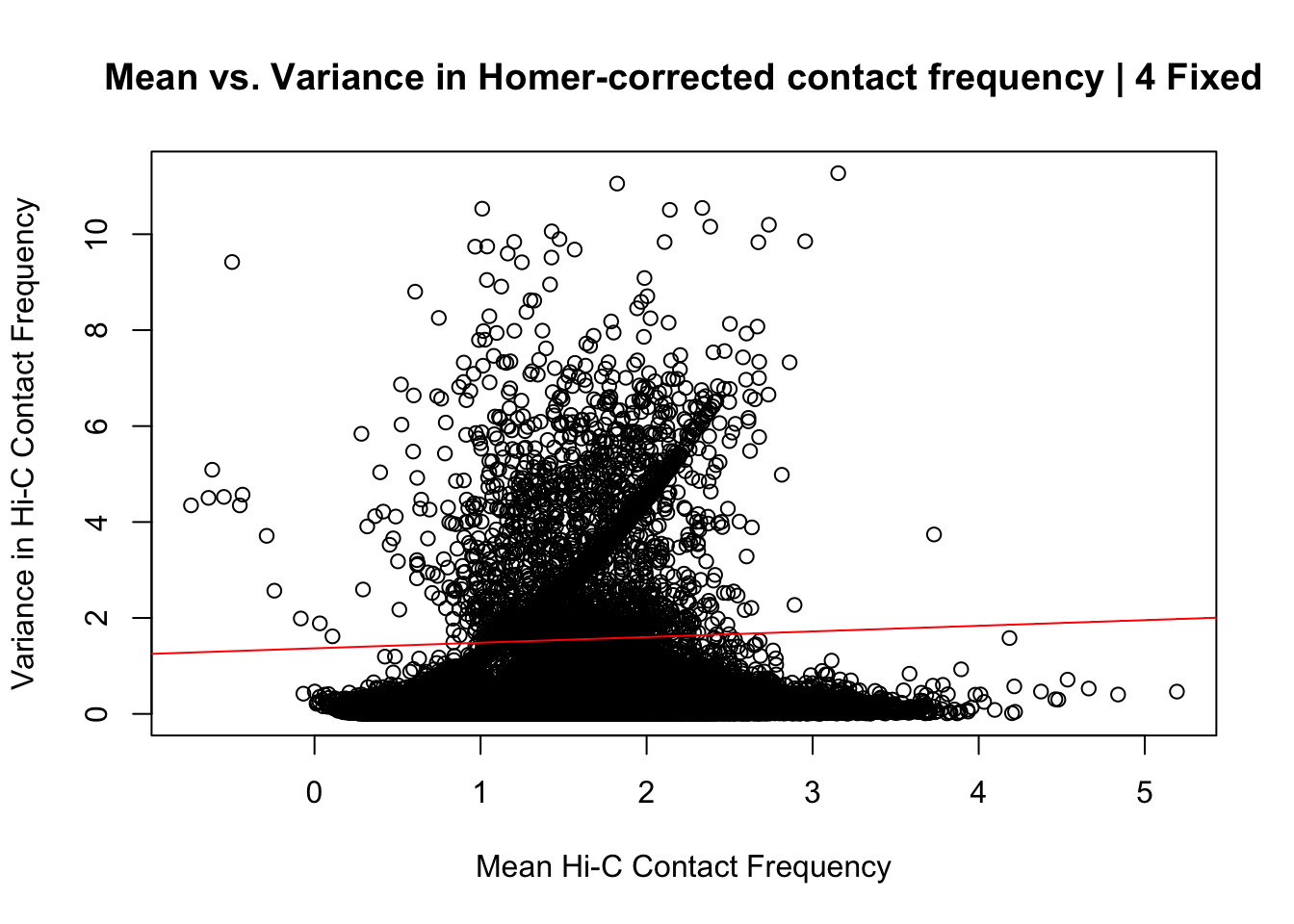
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
summary(lm(data.4.fixed$ALLmean~data.4.fixed$ALLvar))$r.squared[1] 0.008487146summary(lm(data.4.fixed$ALLmean~data.4.fixed$ALLvar))$adj.r.squared[1] 0.008481111#Again, a slight upward trend is seen, but the relationship is quite weak--r squared of 0.008.
###Now to do PCA on the data conditioned upon discovery in 4 individuals--would expect many of the same patterns here, perhaps with even stronger separation due to only grabbing hits that are shared by 4 individuals:
pca4 <- prcomp(t(data.4[,304:311]), scale=TRUE, center=TRUE)
#FIG1A
ggplot(data=as.data.frame(pca4$x), aes(x=PC1, y=PC2, shape=as.factor(meta.data$SP), color=as.factor(meta.data$Batch), size=2)) + geom_point() +labs(title="PCA on Normalized Hi-C Contact Frequency") + guides(color=guide_legend(order=1, title="Batch"), size=FALSE, shape=guide_legend(order=2, title="Species")) + xlab(paste("PC1 (", signif(100*summary(pca4)$importance[2,1],2), "% of variance)")) + ylab((paste("PC2 (", signif(100*summary(pca4)$importance[2,2],2), "% of variance)"))) + labs(color="Batch", shape="Species") + scale_shape_manual(labels=c("Chimp", "Human"), values=c(16, 17)) #Metadata hasn't changed so use the same metadata from above. PCA shows a similar separation along species lines for PC1, with this time it representing 24.171% of the variance (up from 20.49% earlier). This is rather unsurprising, since we have subset down to hits that are more shared than the total set we had before.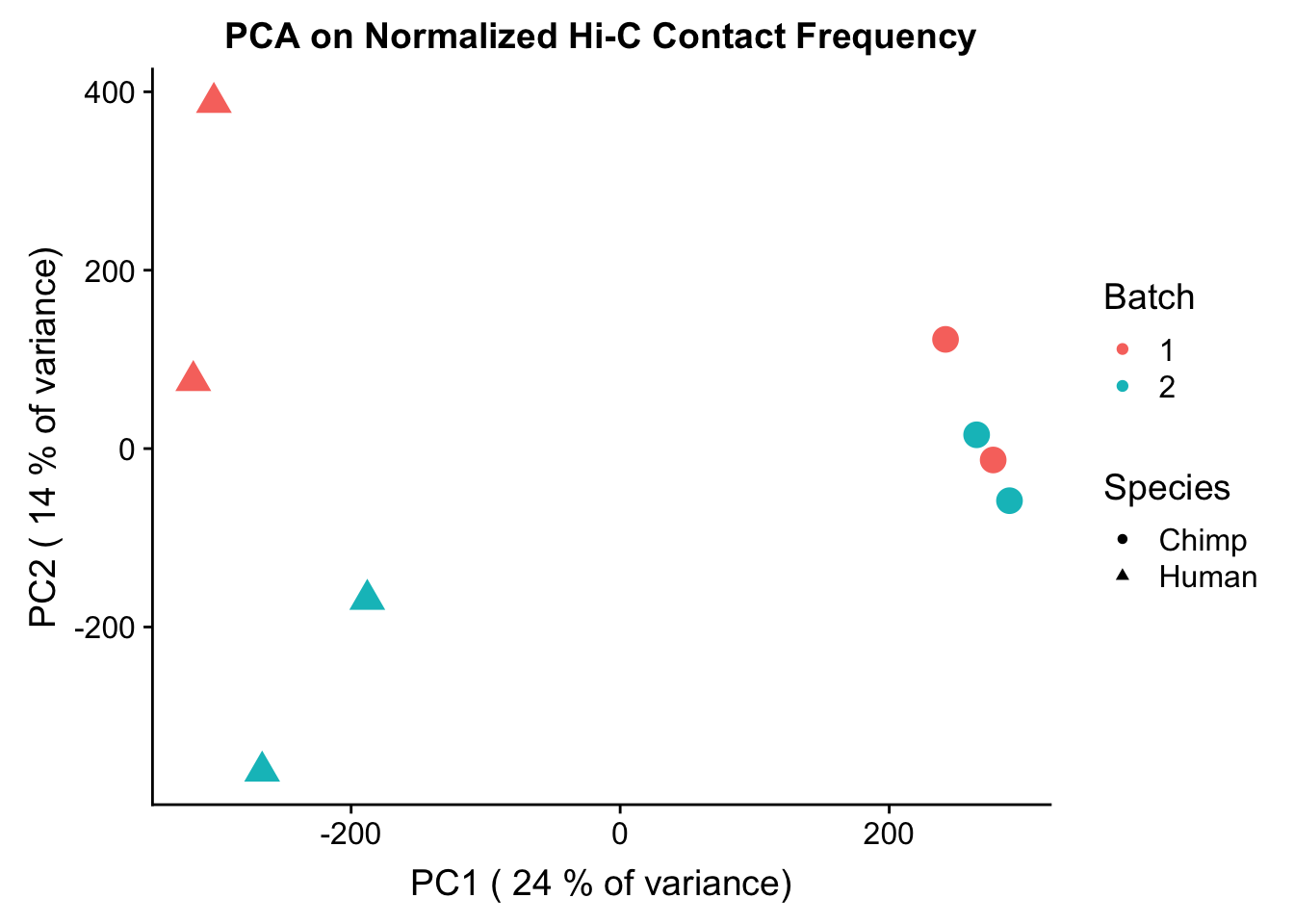
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
FIG1A <- ggplot(data=as.data.frame(pca4$x), aes(x=PC1, y=PC2, shape=as.factor(meta.data$SP), color=as.factor(meta.data$Batch), size=2)) + geom_point() +labs(title="PCA on Normalized Hi-C Contact Frequency") + guides(color=guide_legend(order=1, title="Batch"), size=FALSE, shape=guide_legend(order=2, title="Species")) + xlab(paste("PC1 (", signif(100*summary(pca4)$importance[2,1],2), "% of variance)")) + ylab((paste("PC2 (", signif(100*summary(pca4)$importance[2,2],2), "% of variance)"))) + labs(color="Batch", shape="Species") + scale_shape_manual(labels=c("Chimp", "Human"), values=c(16, 17))
barplot(summary(pca4)$importance[2,], xlab="PCs", ylab="Proportion of Variance Explained", main="PCA on normalized Hi-C contact frequency") #Scree plot showing all the PCs and the proportion of the variance they explain. This is similar to above, with no PCs beyond a 7th one, and the first PC taking the vast majority of the variance while the others hover near 12%. No variables in experimentation, library prep, sequencing batch, or anything else seem to correlate with these PCs.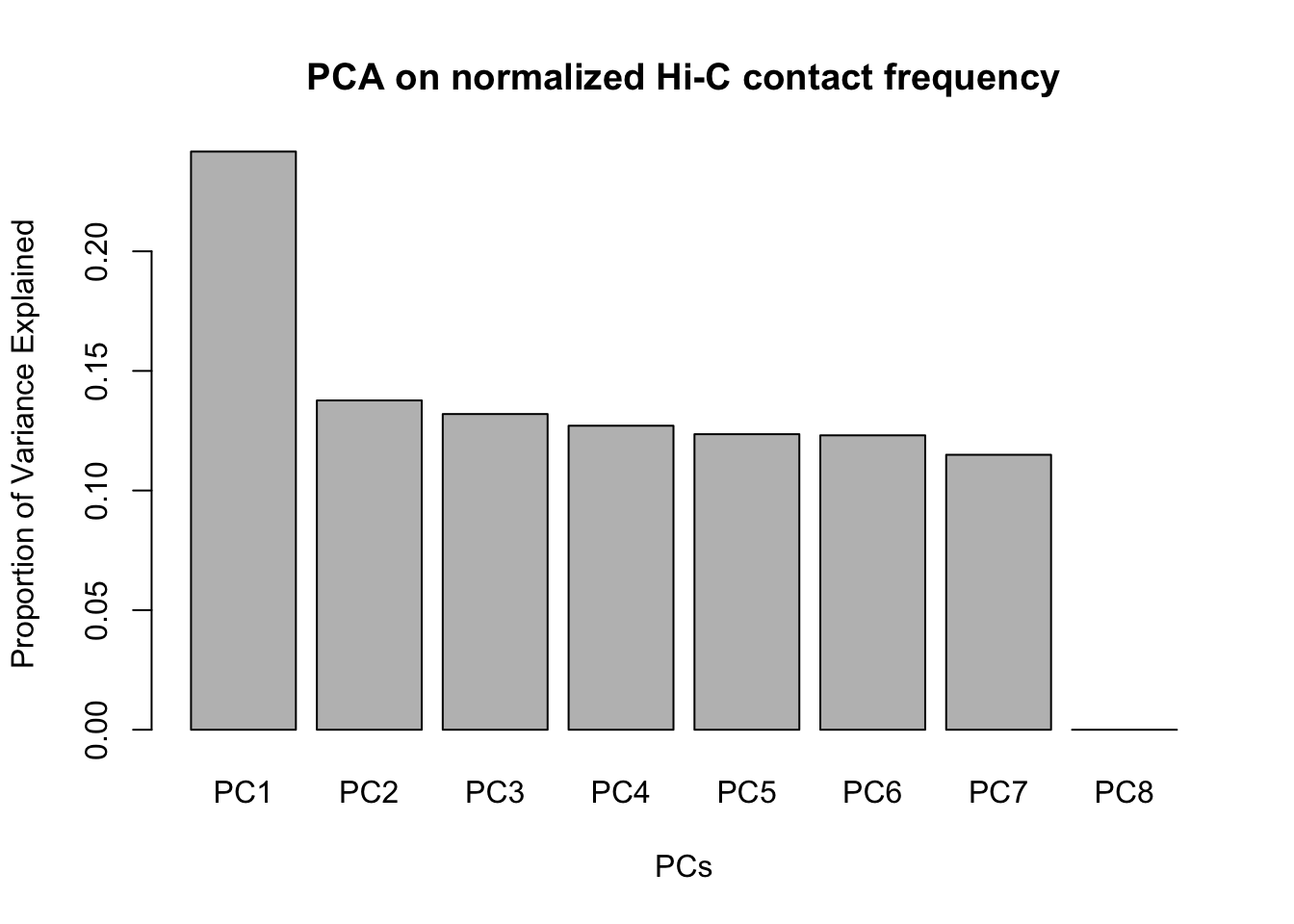
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
plot_grid(FIG1A, labels=c("A"), align="h")#, rel_widths = c(1, 0.01))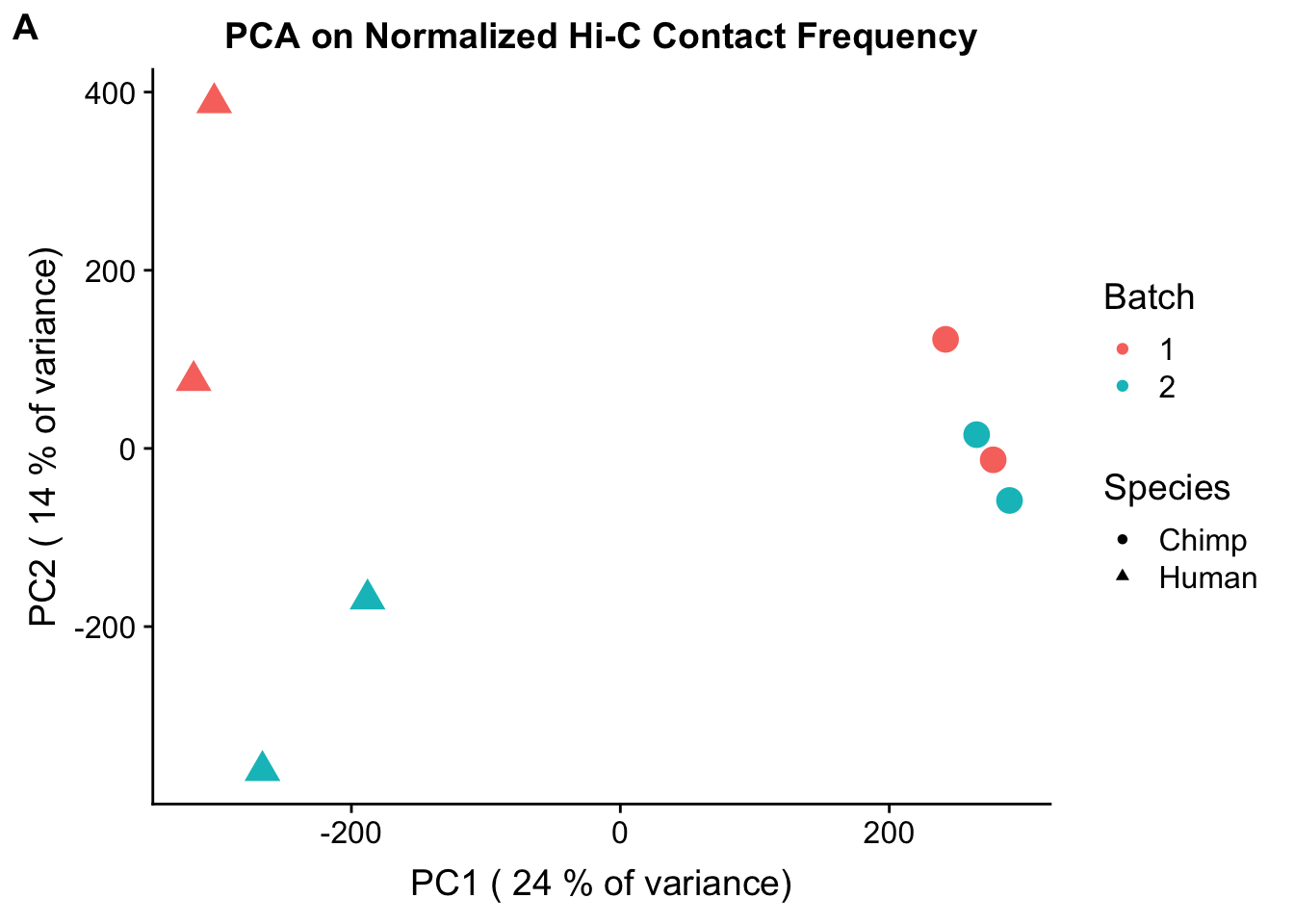
#Actually statistically test correlation w/ PCs:
PC1 <- pca4$x[,1]
PC2 <- pca4$x[,2]
PC3 <- pca4$x[,3]
PC4 <- pca4$x[,4]
PC5 <- pca4$x[,5]
PC6 <- pca4$x[,6]
PC7 <- pca4$x[,7]
PCS <- data.frame(PC1, PC2, PC3, PC4, PC5, PC6, PC7)
summary <- summary(pca4)
PC_pvals <- matrix(data=NA, nrow=7, ncol=6, dimnames=list(c("PC1", "PC2", "PC3", "PC4", "PC5", "PC6", "PC7"), c("Species", "Sex", "Batch", "PE_reads", "tags_per_BP", "inter_reads")))
PC_rsquareds <- matrix(data=NA, nrow=7, ncol=6, dimnames=list(c("PC1", "PC2", "PC3", "PC4", "PC5", "PC6", "PC7"), c("Species", "Sex", "Batch", "PE_reads", "tags_per_BP", "inter_reads")))
for(i in 1:7){
#For species
speciesPC1 <- lm(PCS[,i] ~ as.factor(meta.data$SP))
fstat <- as.data.frame(summary(speciesPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,1] <- p_fstat
PC_rsquareds[i,1] <- summary(speciesPC1)$r.squared
#For sex
sexPC1 <- lm(PCS[,i] ~ as.factor(meta.data$SX))
fstat <- as.data.frame(summary(sexPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,2] <- p_fstat
PC_rsquareds[i,2] <- summary(sexPC1)$r.squared
#For batch
btcPC1 <- lm(PCS[,i] ~ as.factor(meta.data$Batch))
fstat <- as.data.frame(summary(btcPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,3] <- p_fstat
PC_rsquareds[i,3] <- summary(btcPC1)$r.squared
#For PE_reads
PEPC1 <- lm(PCS[,i] ~ meta.data$PE_reads)
fstat <- as.data.frame(summary(PEPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,4] <- p_fstat
PC_rsquareds[i,4] <- summary(PEPC1)$r.squared
#For tags
tagsPC1 <- lm(PCS[,i] ~ meta.data$tags_per_BP)
fstat <- as.data.frame(summary(tagsPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,5] <- p_fstat
PC_rsquareds[i,5] <- summary(tagsPC1)$r.squared
#For inter
interPC1 <- lm(PCS[,i] ~ meta.data$inter_reads)
fstat <- as.data.frame(summary(interPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,6] <- p_fstat
PC_rsquareds[i,6] <- summary(interPC1)$r.squared
}
PC_pvals Species Sex Batch PE_reads tags_per_BP inter_reads
PC1 2.199084e-06 0.89410140 0.82762305 0.08306967 0.4162871 0.89012226
PC2 8.476933e-01 0.63365009 0.05252363 0.28149929 0.1689697 0.05773082
PC3 8.009030e-01 0.62475973 0.48718261 0.28722360 0.1654083 0.93235638
PC4 9.476353e-01 0.02066748 0.66273167 0.98208331 0.9974648 0.67607958
PC5 9.823623e-01 0.75209783 0.28215075 0.39670984 0.2933573 0.31892644
PC6 9.873455e-01 0.24571167 0.96034286 0.80290246 0.7561813 0.36092781
PC7 9.816176e-01 0.55255454 0.27645287 0.51392897 0.4313247 0.29167386PC_rsquareds Species Sex Batch PE_reads tags_per_BP
PC1 9.808831e-01 0.003203562 0.0085488465 4.182812e-01 1.126790e-01
PC2 6.657192e-03 0.040285292 0.4920198755 1.891727e-01 2.894772e-01
PC3 1.144877e-02 0.042382922 0.0836525906 1.851745e-01 2.935635e-01
PC4 7.807753e-04 0.618194228 0.0338509794 9.131995e-05 1.828203e-06
PC5 8.849812e-05 0.017903221 0.1887135525 1.218537e-01 1.809798e-01
PC6 4.555294e-05 0.216177784 0.0004476097 1.121650e-02 1.730444e-02
PC7 9.612991e-05 0.061852991 0.1927665459 7.421000e-02 1.059942e-01
inter_reads
PC1 0.003449966
PC2 0.477456646
PC3 0.001303785
PC4 0.031110479
PC5 0.164427326
PC6 0.140129734
PC7 0.182122065#So PC1 correlates strongly and significantly with species, whereas PC4 has some moderate correlation with sex--but nothing else has a significant effect.
###Heatmap clustering of data after conditioning upon discovery in 4 individuals (regardless of species). Would expect to see the same clustering, with perhaps higher correlation values due to conditioning upon discovery in 4 individuals.
corheat4 <- cor(data.4[,304:311], use="complete.obs", method="pearson") #Corheat for the full data set, and heatmap
#colnames(corheat4) <- c("A_HF", "B_HM", "C_CM", "D_CF", "E_HM", "F_HF", "G_CM", "H_CF")
colnames(corheat4) <- c("H_F1", "H_M1", "C_M1", "C_F1", "H_M2", "H_F2", "C_M2", "C_F2") #Better for presentation
rownames(corheat4) <- colnames(corheat4)
#Change path to make sure orca can be found (in conda path); set mapbox token for orca export.
Sys.setenv(PATH="/usr/bin:/bin:/usr/sbin:/sbin:/usr/local/bin:/opt/X11/bin:/Library/TeX/texbin:/opt/local/bin:/Users/ittaieres/miniconda3/bin/")
Sys.setenv('MAPBOX_TOKEN'='pk.eyJ1IjoiaXR0YWllcmVzIiwiYSI6ImNqeHVwanphejE3bjIzcHFmY2FvYXIxZXUifQ.rs3I8LoxJcqWBsIhVDiJCQ')
#FIG1B
heatmaply(corheat4, main="Pairwise Pearson Correlation Clustering @ 10kb", k_row=2, k_col=2, symm=TRUE, margins=c(50, 50, 30, 30), cellnote_size=10, fontsize_row=10, fontsize_col=10, file="~/Desktop/Fig1B.pdf")#, scale_fill_gradient_fun = ggplot2::scale_fill_gradient2(low = "yellow", high = "blue", midpoint = 0.5, limits = c(0, 1)))dis <- heatmaply(corheat4, main="Pairwise Pearson Correlation Clustering @ 10kb", k_row=2, k_col=2, symm=TRUE, margins=c(50, 50, 30, 30), cellnote_size=10, fontsize_row=10, fontsize_col=10)
orca(dis, file="~/Desktop/Fig1B.pdf") #Note that this just created a ~ folder in the hicipsc directory, didn't redirect to home folder.
#Indeed, we can again see that all the humans cluster together, as do all the chimps. This is very good, and given this new condition, we also see higher within-species correlations for the significant hits, in the 0.6-0.7 range (was much closer to 0.3 before).
FIG1B <- heatmaply(corheat4, main="Pairwise Pearson Correlation Clustering @ 10kb", k_row=2, k_col=2, symm=TRUE, margins=c(50, 50, 30, 30), plot_method = "ggplot", return_ppxpy = TRUE)
#FIG1B <- readPNG("~/Downloads/newplot (16).png")
#theme_cowplot(font_size=10, font_family="Arial")
#Need to mess with this now to get it paper-ready:
# FIG1B <- FIG1B[!sapply(FIG1B, is.null)]
# FIG1B <- lapply(FIG1B, ggplotGrob)
# FIG1B$p$widths <- FIG1B$px$widths <- FIG1B$py$widths <- unit.pmax(
# FIG1B$p$widths,
# FIG1B$px$widths,
# FIG1B$py$widths)
#
# FIG1B$p$heights <- FIG1B$px$heights <- FIG1B$py$heights <- unit.pmax(
# FIG1B$p$heights,
# FIG1B$px$heights,
# FIG1B$py$heights)
#
# grid.arrange(FIG1B$py, textGrob(""), FIG1B$p, FIG1B$px, nrow=2)
#
# FIG1 <- plot_grid(FIG1A, labels=c("B", "B"), align="h")#, rel_widths = c(1, 0.01))
# save_plot("~/Desktop/test.eps", FIG1)
#
#
# save_plot("~/Desktop/Paper Drafts/PLOS/Revision/Revision_2/Final?/FINAL/figures/FIGS8A.eps", FIGS8A)
###Repeat the above, but conditioned upon discovery in 4 individuals within a species--would expect many of the same patterns here, this time with much stronger separation due to only grabbing hits that are fixed in a species:
pca4 <- prcomp(t(data.4.fixed[,304:311]), scale=TRUE, center=TRUE)
ggplot(data=as.data.frame(pca4$x), aes(x=PC1, y=PC2, shape=as.factor(meta.data$SP), color=as.factor(meta.data$Batch), size=2)) + geom_point() +labs(title="PCA on Normalized Hi-C Contact Frequency") + guides(color=guide_legend(order=1, title="Batch"), size=FALSE, shape=guide_legend(order=2, title="Species")) + xlab(paste("PC1 (", signif(100*summary(pca4)$importance[2,1],2), "% of variance)")) + ylab((paste("PC2 (", signif(100*summary(pca4)$importance[2,2],2), "% of variance)"))) + labs(color="Batch", shape="Species") + scale_shape_manual(labels=c("Chimp", "Human"), values=c(16, 17)) #Metadata hasn't changed so use the same metadata from above. PCA shows a similar separation along species lines for PC1, with this time it representing 24.171% of the variance (up from 20.49% earlier). This is rather unsurprising, since we have subset down to hits that are more shared than the total set we had before.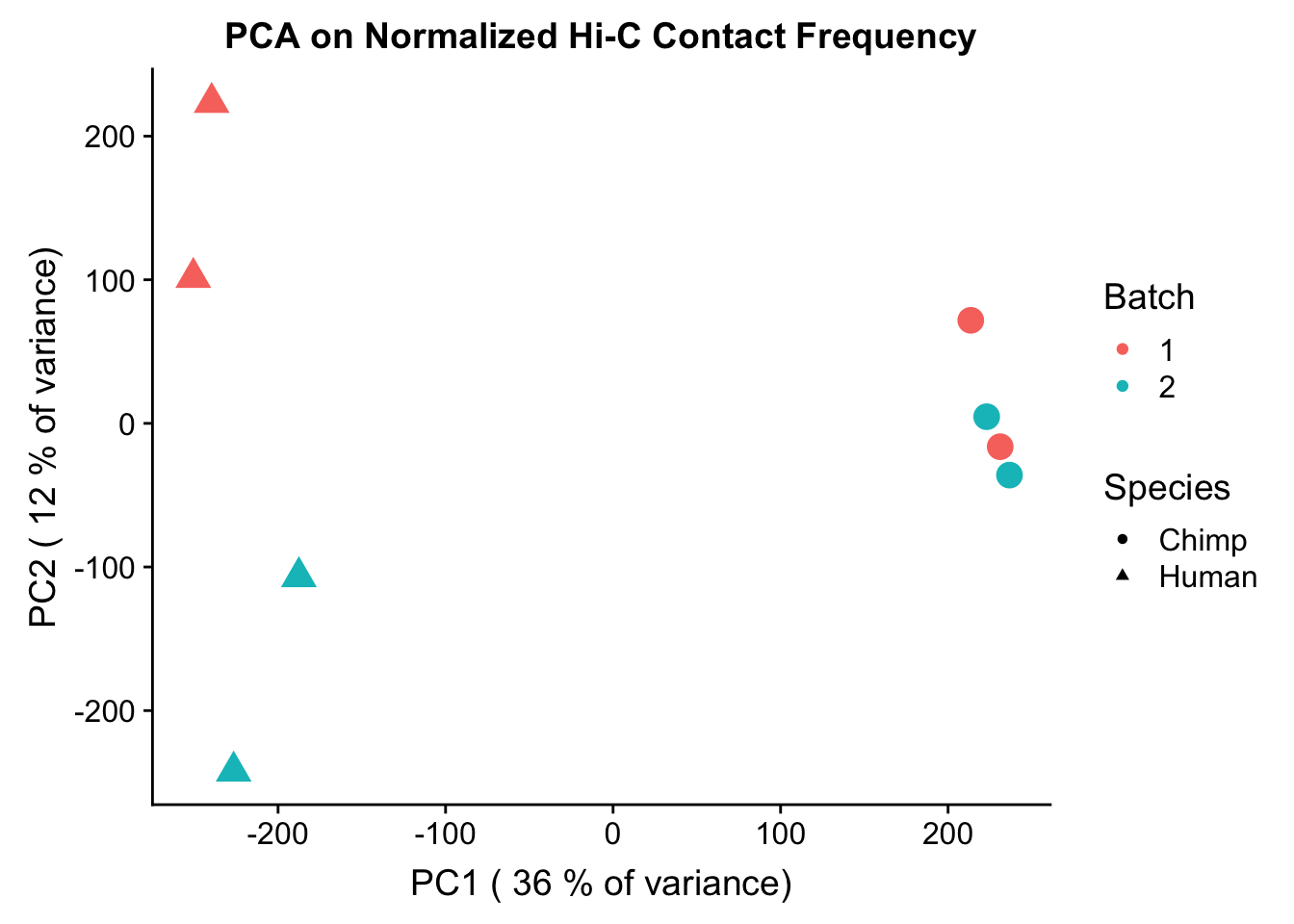
| Version | Author | Date |
|---|---|---|
| 2ec8067 | Ittai Eres | 2019-05-09 |
barplot(summary(pca4)$importance[2,], xlab="PCs", ylab="Proportion of Variance Explained", main="PCA on normalized Hi-C contact frequency") #Scree plot showing all the PCs and the proportion of the variance they explain. This is similar to above, with no PCs beyond a 7th one, and the first PC taking the vast majority of the variance while the others hover near 12%. No variables in experimentation, library prep, sequencing batch, or anything else seem to correlate with these PCs.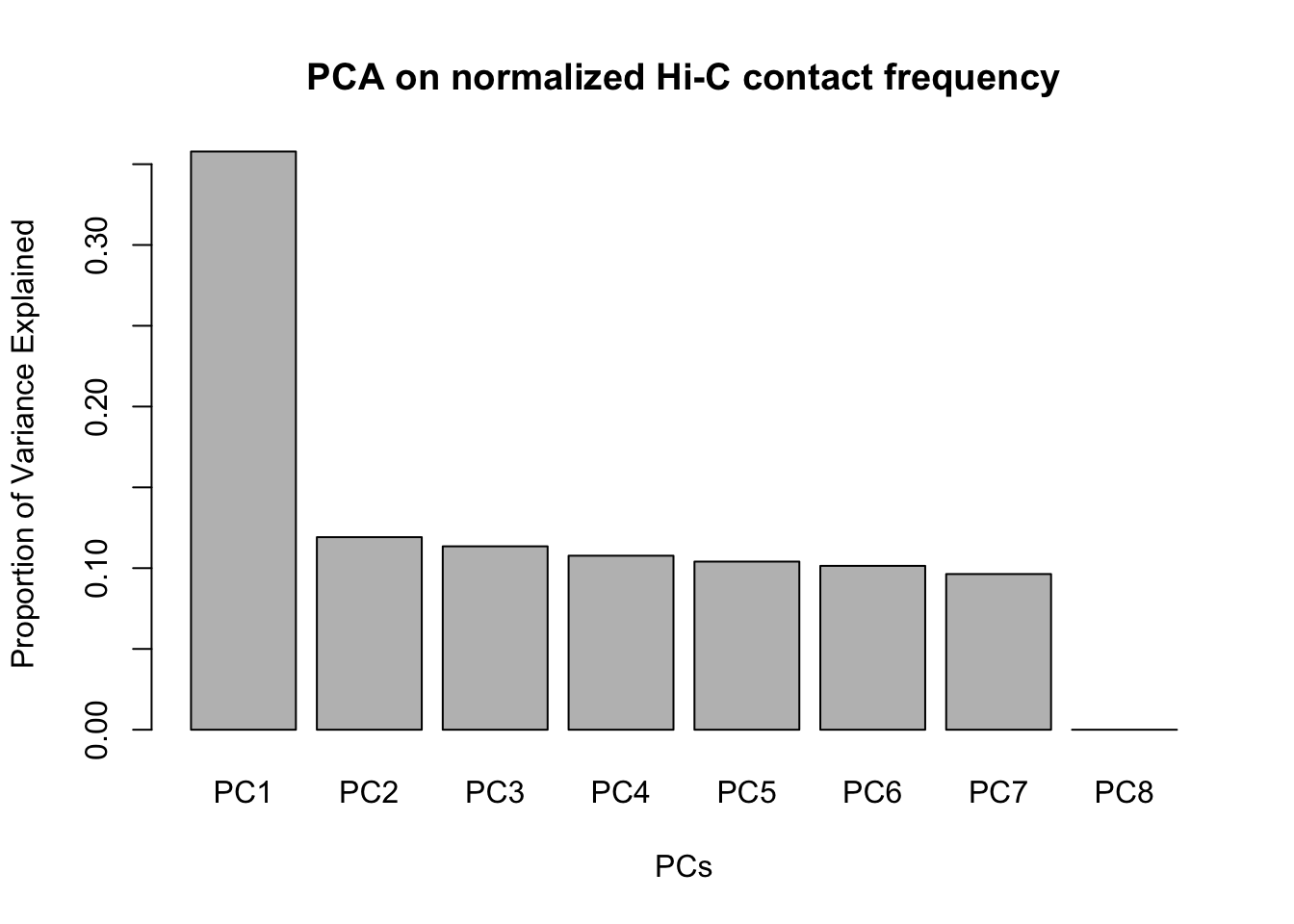
#Actually statistically test correlation w/ PCs:
PC1 <- pca4$x[,1]
PC2 <- pca4$x[,2]
PC3 <- pca4$x[,3]
PC4 <- pca4$x[,4]
PC5 <- pca4$x[,5]
PC6 <- pca4$x[,6]
PC7 <- pca4$x[,7]
PCS <- data.frame(PC1, PC2, PC3, PC4, PC5, PC6, PC7)
summary <- summary(pca4)
PC_pvals <- matrix(data=NA, nrow=7, ncol=6, dimnames=list(c("PC1", "PC2", "PC3", "PC4", "PC5", "PC6", "PC7"), c("Species", "Sex", "Batch", "PE_reads", "tags_per_BP", "inter_reads")))
PC_rsquareds <- matrix(data=NA, nrow=7, ncol=6, dimnames=list(c("PC1", "PC2", "PC3", "PC4", "PC5", "PC6", "PC7"), c("Species", "Sex", "Batch", "PE_reads", "tags_per_BP", "inter_reads")))
for(i in 1:7){
#For species
speciesPC1 <- lm(PCS[,i] ~ as.factor(meta.data$SP))
fstat <- as.data.frame(summary(speciesPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,1] <- p_fstat
PC_rsquareds[i,1] <- summary(speciesPC1)$r.squared
#For sex
sexPC1 <- lm(PCS[,i] ~ as.factor(meta.data$SX))
fstat <- as.data.frame(summary(sexPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,2] <- p_fstat
PC_rsquareds[i,2] <- summary(sexPC1)$r.squared
#For batch
btcPC1 <- lm(PCS[,i] ~ as.factor(meta.data$Batch))
fstat <- as.data.frame(summary(btcPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,3] <- p_fstat
PC_rsquareds[i,3] <- summary(btcPC1)$r.squared
#For PE_reads
PEPC1 <- lm(PCS[,i] ~ meta.data$PE_reads)
fstat <- as.data.frame(summary(PEPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,4] <- p_fstat
PC_rsquareds[i,4] <- summary(PEPC1)$r.squared
#For tags
tagsPC1 <- lm(PCS[,i] ~ meta.data$tags_per_BP)
fstat <- as.data.frame(summary(tagsPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,5] <- p_fstat
PC_rsquareds[i,5] <- summary(tagsPC1)$r.squared
#For inter
interPC1 <- lm(PCS[,i] ~ meta.data$inter_reads)
fstat <- as.data.frame(summary(interPC1)$fstatistic)
p_fstat <- 1-pf(fstat[1,], fstat[2,], fstat[3,])
PC_pvals[i,6] <- p_fstat
PC_rsquareds[i,6] <- summary(interPC1)$r.squared
}
PC_pvals Species Sex Batch PE_reads tags_per_BP inter_reads
PC1 7.652116e-08 0.9388975 0.90588992 0.1101099 0.4897443 0.84521168
PC2 9.137237e-01 0.7953693 0.04114024 0.2299095 0.1372477 0.05787434
PC3 8.822923e-01 0.4996362 0.57917517 0.2934863 0.1894047 0.87875726
PC4 9.808643e-01 0.0688759 0.76645999 0.9397405 0.9313555 0.72218379
PC5 9.870802e-01 0.1357245 0.55304458 0.4402560 0.3371011 0.20514700
PC6 9.994307e-01 0.6446713 0.35595387 0.5976319 0.5247353 0.64308606
PC7 9.928907e-01 0.4713467 0.27360567 0.5320021 0.4561680 0.26751028PC_rsquareds Species Sex Batch PE_reads tags_per_BP
PC1 9.937487e-01 0.001063486 0.002527747 0.36913936 0.082715030
PC2 2.123294e-03 0.012104623 0.528109515 0.22934399 0.328843691
PC3 3.961882e-03 0.079161470 0.054151272 0.18089261 0.267396647
PC4 1.041708e-04 0.449366816 0.015845287 0.00103430 0.001342725
PC5 4.748306e-05 0.330921761 0.061705395 0.10216537 0.153502492
PC6 9.218845e-08 0.037770741 0.142837286 0.04917722 0.070605503
PC7 1.437685e-05 0.089611103 0.194823498 0.06824714 0.095593912
inter_reads
PC1 0.006877925
PC2 0.477069591
PC3 0.004204788
PC4 0.022626868
PC5 0.251794032
PC6 0.038126629
PC7 0.199300166#Once again, PC1 correlates strongly and significantly with species, and this time PC4 has non-significant moderate correlation with sex. PC2 has a stastically significant but fairly weak r squared wiht batch, and nothing else has a significant effect.
###Heatmap clustering of data after conditioning upon discovery in 4 individuals (regardless of species). Would expect to see the same clustering, with perhaps higher correlation values due to conditioning upon discovery in 4 individuals.
corheat4 <- cor(data.4[,304:311], use="complete.obs", method="pearson") #Corheat for the full data set, and heatmap
#colnames(corheat4) <- c("A_HF", "B_HM", "C_CM", "D_CF", "E_HM", "F_HF", "G_CM", "H_CF")
colnames(corheat4) <- c("H_F1", "H_M1", "C_M1", "C_F1", "H_M2", "H_F2", "C_M2", "C_F2") #Better for presentation
rownames(corheat4) <- colnames(corheat4)
#Here I just make a fake PCA plot with some words on it so I can use them as labels on the heatmaply plot (for consistency)
ggplot(data=as.data.frame(pca4$x), aes(x=PC1, y=PC2, shape=as.factor(meta.data$SP), color=as.factor(meta.data$Batch), size=2)) + geom_point() +labs(title="Pairwise Pearson Correlation Clustering @ 10kb") + guides(color=guide_legend(order=1, title="Pearson Correlation"), size=FALSE, shape=guide_legend(order=2, title="Species")) + xlab(paste("PC1 (", signif(100*summary(pca4)$importance[2,1],2), "% of variance)")) + ylab((paste("PC2 (", signif(100*summary(pca4)$importance[2,2],2), "% of variance)"))) + labs(color="Batch", shape="Species") + scale_shape_manual(labels=c("Chimp", "Human"), values=c(16, 17))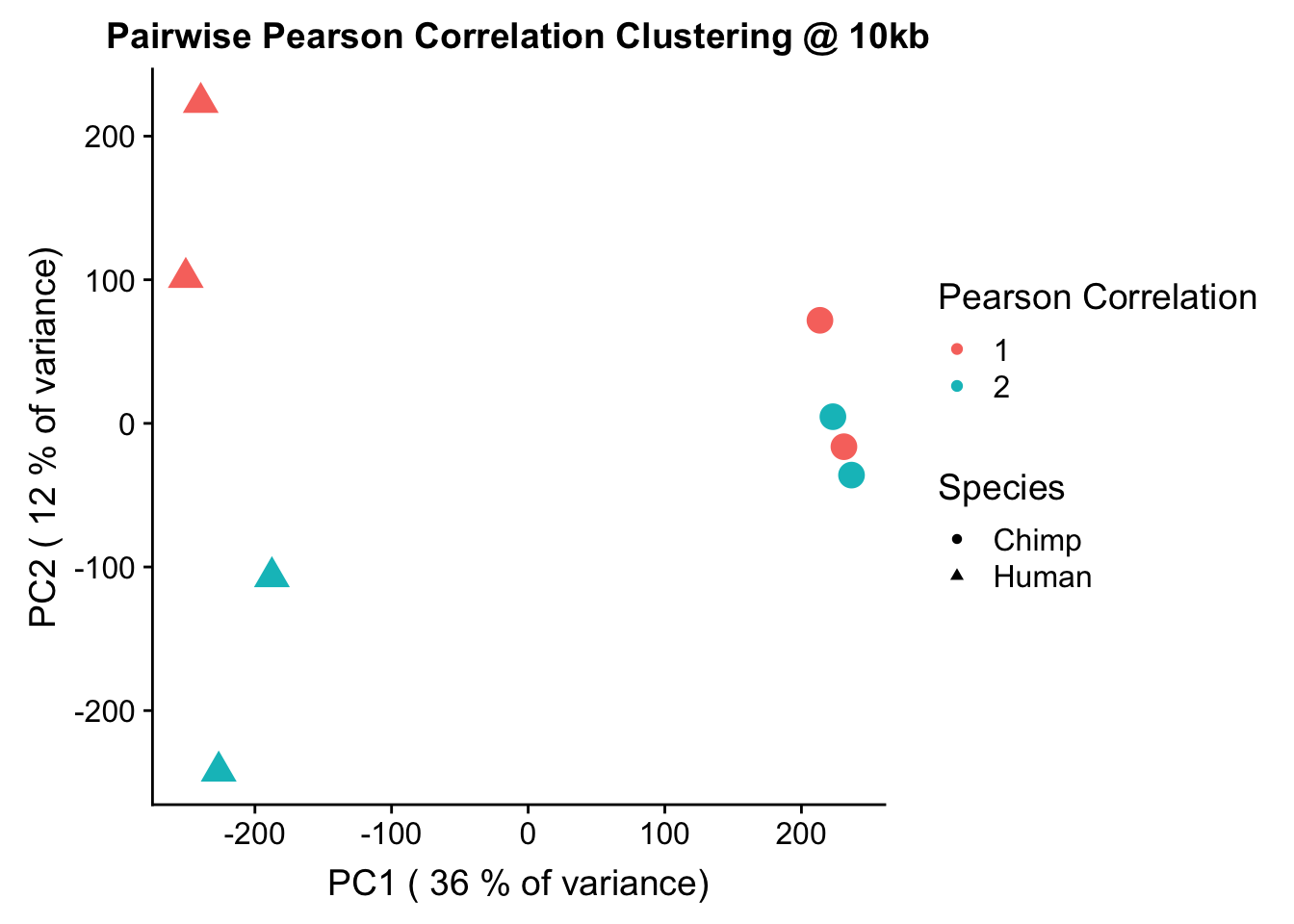
| Version | Author | Date |
|---|---|---|
| 2ec8067 | Ittai Eres | 2019-05-09 |
#Repeat all this for data conditioned upon a fixed significant Hi-C contact within a species:
corheat4 <- cor(data.4.fixed[,304:311], use="complete.obs", method="pearson") #Corheat for the full data set, and heatmap
#colnames(corheat4) <- c("A_HF", "B_HM", "C_CM", "D_CF", "E_HM", "F_HF", "G_CM", "H_CF")
colnames(corheat4) <- c("HF1", "HM1", "CM1", "CF1", "HM2", "HF2", "CM2", "CF2") #Better for presentation
rownames(corheat4) <- colnames(corheat4)
heatmaply(corheat4, main="Pairwise Pearson Correlation Clustering @ 10kb | 4 Species", k_row=2, k_col=2, symm=TRUE, margins=c(50, 50, 30, 30))#, scale_fill_gradient_fun = ggplot2::scale_fill_gradient2(low = "yellow", high = "blue", midpoint = 0.5, limits = c(0, 1)))#Indeed, we can again see that all the humans cluster together, as do all the chimps. This is very good, and given this new condition, we also see higher within-species correlations for the significant hits, in the 0.6-0.7 range (was much closer to 0.3 before).
#Here I just make a fake PCA plot with some words on it so I can use them as labels on the heatmaply plot (for consistency)
ggplot(data=as.data.frame(pca4$x), aes(x=PC1, y=PC2, shape=as.factor(meta.data$SP), color=as.factor(meta.data$Batch), size=2)) + geom_point() +labs(title="Pairwise Pearson Correlation Clustering @ 10kb") + guides(color=guide_legend(order=1, title="Pearson Correlation"), size=FALSE, shape=guide_legend(order=2, title="Species")) + xlab(paste("PC1 (", signif(100*summary(pca4)$importance[2,1],2), "% of variance)")) + ylab((paste("PC2 (", signif(100*summary(pca4)$importance[2,2],2), "% of variance)"))) + labs(color="Batch", shape="Species") + scale_shape_manual(labels=c("Chimp", "Human"), values=c(16, 17))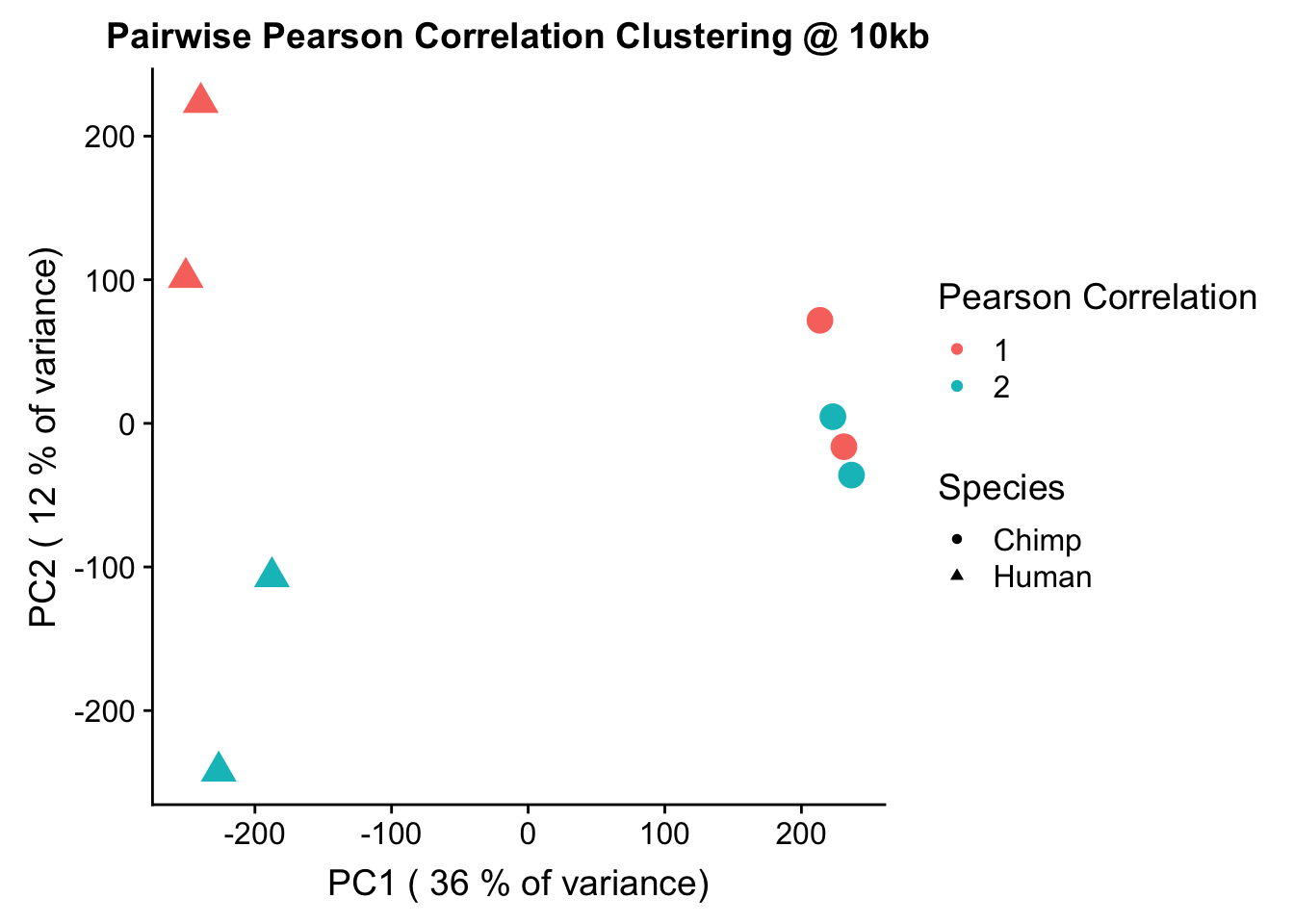
| Version | Author | Date |
|---|---|---|
| 2ec8067 | Ittai Eres | 2019-05-09 |
###This initial QC suggest quality of the data is high, and has given us some information on how to filter the data before further intrepretation. When working locally, can be helpful to write out both the full dataset and the one conditioned upon 4 individuals, in order to do sanity checks on both later on. Note that a call of identical on something fread in after fwriting it out will still return FALSE with this as floating point can't represent decimals exactly (see https://stackoverflow.com/questions/9508518/why-are-these-numbers-not-equal or https://cran.r-project.org/doc/FAQ/R-FAQ.html#Why-doesn_0027t-R-think-these-numbers-are-equal_003f):
fwrite(full.data, "output/full.data.10.init.QC", quote = TRUE, sep = "\t", row.names = FALSE, col.names = TRUE, na="NA", showProgress = FALSE)
fwrite(data.4, "output/data.4.init.QC", quote=FALSE, sep="\t", row.names=FALSE, col.names=TRUE, na="NA", showProgress = FALSE)
fwrite(data.4.fixed, "output/data.4.fixed.init.QC", quote=FALSE, sep="\t", row.names=FALSE, col.names=TRUE, na="NA", showProgress = FALSE)Homer-specific QC
#Read in pre-prepped tables of this information.
rand.frag <- fread("~/Desktop/Hi-C/homer_QCs/petag.Local.ALL.distn", header=TRUE, stringsAsFactors = FALSE, data.table=FALSE)
dist.distn <- fread("~/Desktop/Hi-C/homer_QCs/petag.Freq.ALL.distn", header=TRUE, stringsAsFactors = FALSE, data.table=FALSE)
#Create a huge long-form data frame of the same and opposite strand counts from each individual to place in a long-form dataframe for ggplot2 with the local distances and sample identities.
myfrags <- rbind(rand.frag[,2:3], rand.frag[,4:5], rand.frag[,6:7], rand.frag[,8:9], rand.frag[,10:11], rand.frag[,12:13], rand.frag[,14:15], rand.frag[,16:17])
fragdf <- data.frame(local_dist=rand.frag[,1], same=myfrags[,1], opposite=myfrags[,2], ID=c(rep("A_H", 20000), rep("B_H", 20000), rep("C_C", 20000), rep("D_C", 20000), rep("E_H", 20000), rep("F_H", 20000), rep("G_C", 20000), rep("H_C", 20000)))
ggplot(data=fragdf, aes(x=local_dist, group=ID)) + geom_line(aes(y=same, color=ID)) + ggtitle("Relationship b/t 5' ends of paired reads, same strand") + xlab("Local Distance from 5' of 1st read") + ylab("Counts per bp")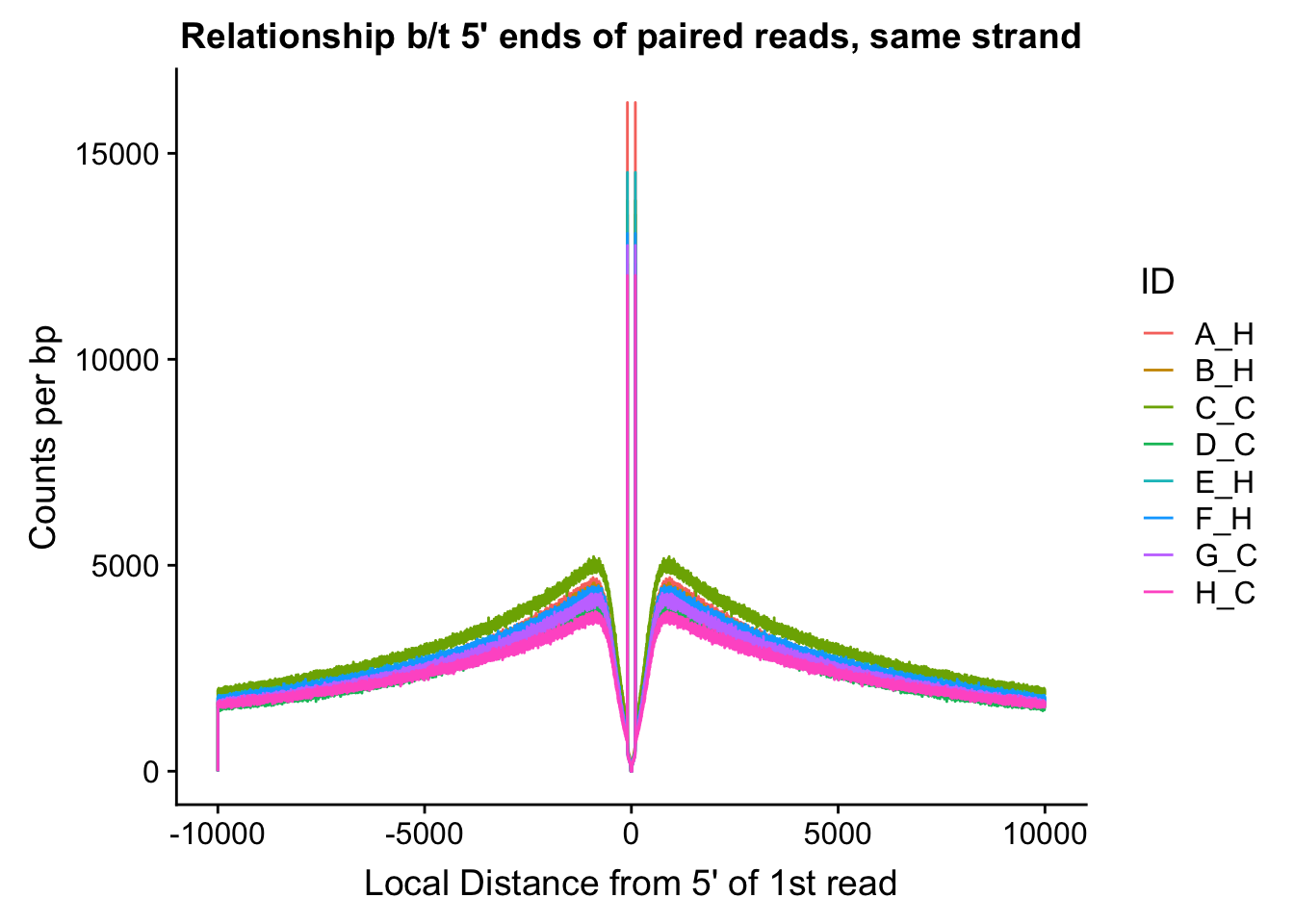
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
ggplot(data=fragdf, aes(x=local_dist, group=ID)) + geom_line(aes(y=opposite, color=ID)) + ggtitle("Relationship b/t 5' ends of paired reads, opposite strand") + xlab("Local Distance from 5' of 1st read") + ylab("Counts per bp")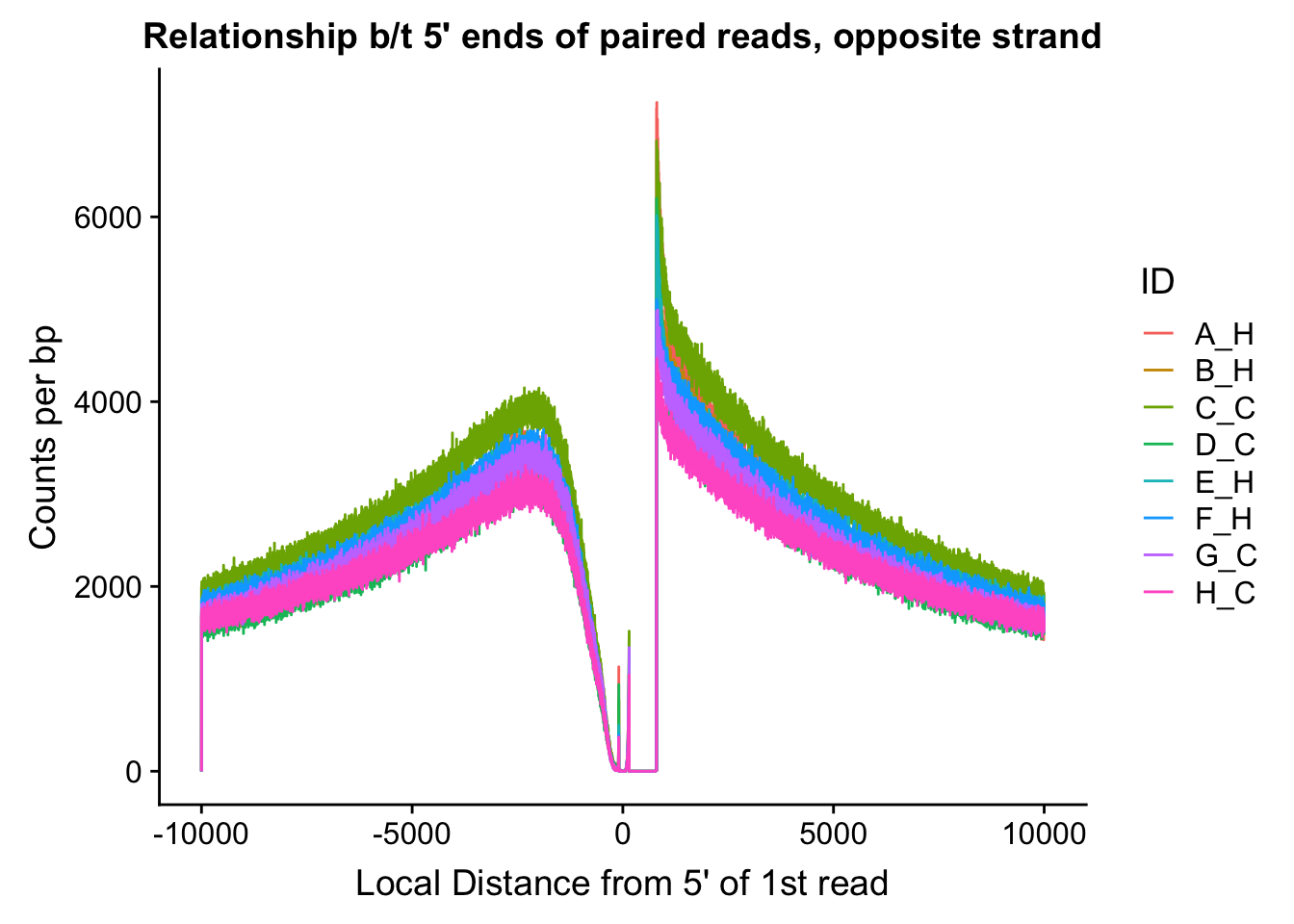
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
#Critically, there appears to be no clear separation between species here. If there is any effect, it would appear to be that of batch, but even this effect appears minor, and would likely be more related to total sequencing reads (batch 1 had the additional pooled lane) than to actual library prep differences.
#Rearrange data on fraction of reads from each library falling within a certain distance of one another so that it can be plotted for all individuals simultaneously in ggplot2.
dist.distn$`Distance between PE tags`[300001] <- 300000000
dist.distn$binID <- c(rep(seq(0, 299999000, 10000), each=10), 300000000)
colnames(dist.distn) <- c("distance", "A_H", "B_H", "C_C", "D_C", "E_H", "F_H", "G_C", "H_C", "binID")
#Group by to lump 10k together at a time.
group_by(dist.distn, binID) %>% summarise(A_H=sum(A_H), B_H=sum(B_H), C_C=sum(C_C), D_C=sum(D_C), E_H=sum(E_H), F_H=sum(F_H), G_C=sum(G_C), H_C=sum(H_C)) -> mydist
#Prepare long-form df of this information for plotting with ggplot.
distdf <- data.frame(dist=rep(mydist$binID, 8), fraction=c(mydist$A_H, mydist$B_H, mydist$C_C, mydist$D_C, mydist$E_H, mydist$F_H, mydist$G_C, mydist$H_C), ID=c(rep("A-21792", 30001), rep("B-28126", 30001), rep("C-3649", 30001), rep("D-40300", 30001), rep("E-28815", 30001), rep("F-28834", 30001), rep("G-3624", 30001), rep("H-3651", 30001)), species=c(rep("Human", 60002), rep("Chimpanzee", 60002), rep("Human", 60002), rep("Chimpanzee", 60002)))
#Now, plot out on several different distance scales to see if there is any strong species effect.
ggplot(data=distdf, aes(x=dist, group=ID)) + geom_line(aes(y=fraction, color=ID, linetype=species)) + coord_cartesian(xlim=c(0, 50000)) + xlab("Distance Between Mates") + ylab("Fraction of Hi-C PE reads") + ggtitle("Hi-C Pair Distance Distributions") #FIGS11B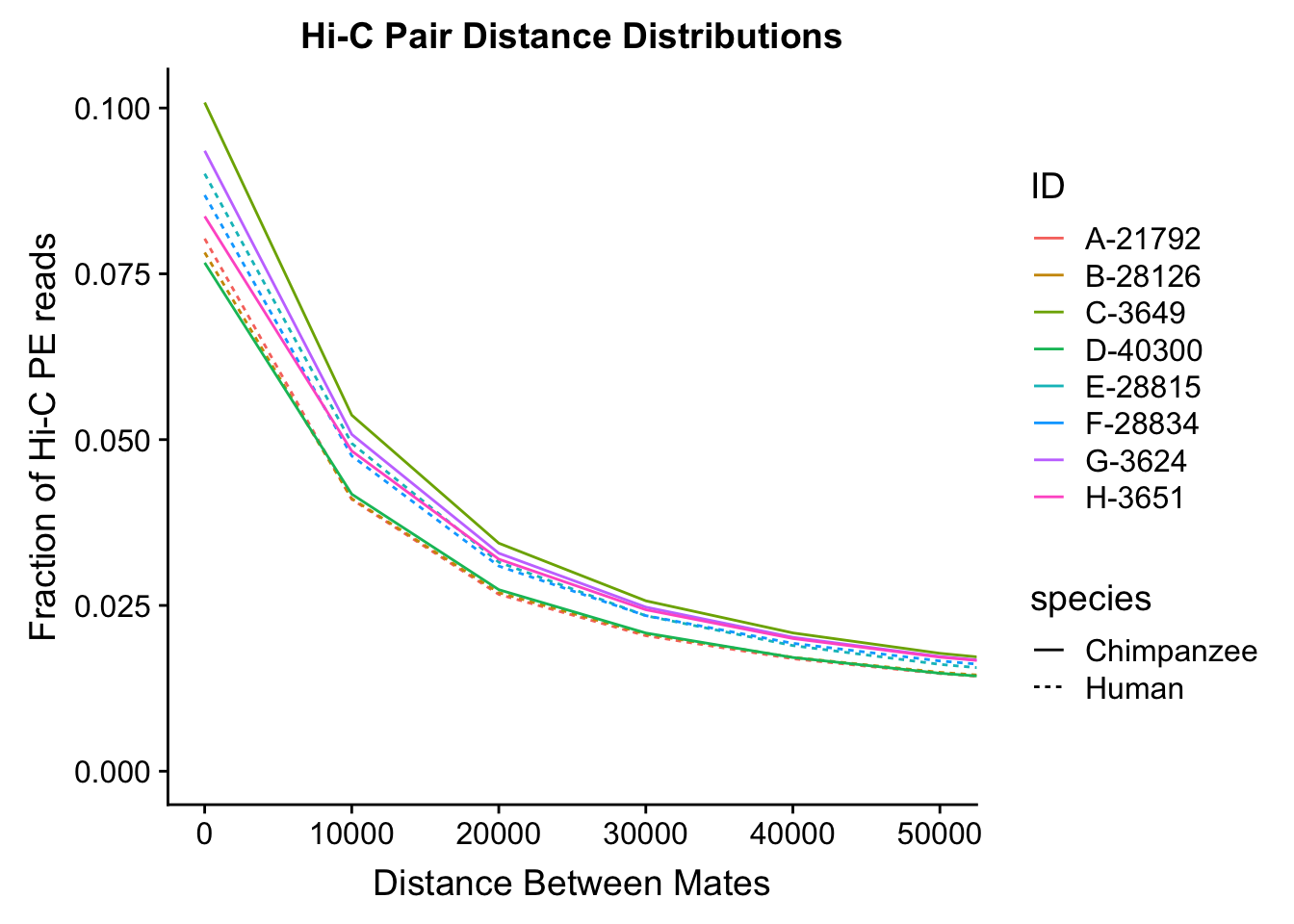
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
ggplot(data=distdf, aes(x=dist, group=ID)) + geom_line(aes(y=fraction, color=ID, linetype=species)) + coord_cartesian(xlim=c(0, 100000)) + xlab("Distance between mates") + ylab("Fraction of Hi-C PE reads") + ggtitle("Hi-C Pair Distance Distributions")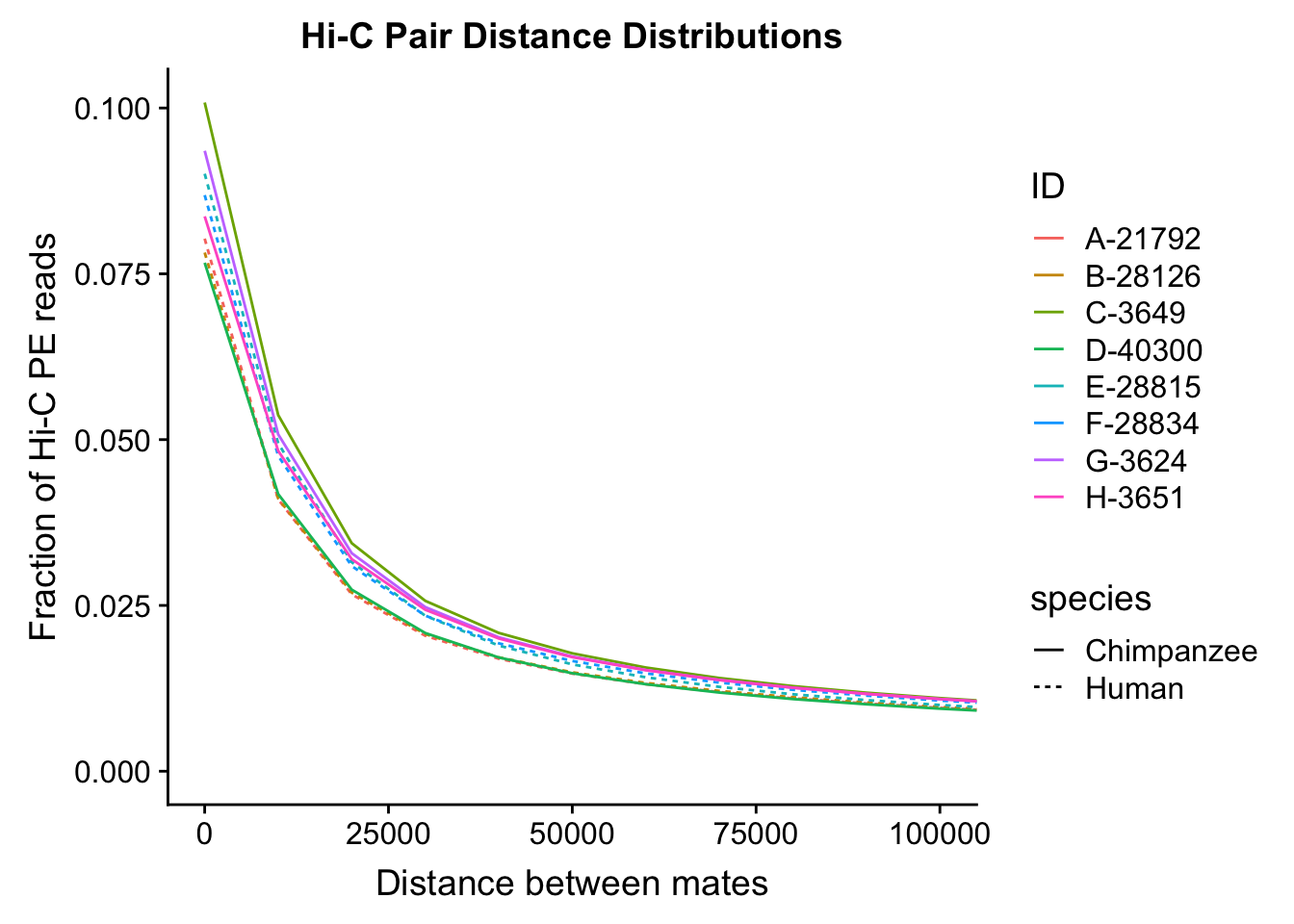
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
ggplot(data=distdf, aes(x=dist, group=ID)) + geom_line(aes(y=fraction, color=ID, linetype=species)) + coord_cartesian(xlim=c(0, 500000)) + xlab("Distance Between mates") + ylab("Fraction of Hi-C PE reads") + ggtitle("Hi-C Pair Distance Distributions") #FIGS11A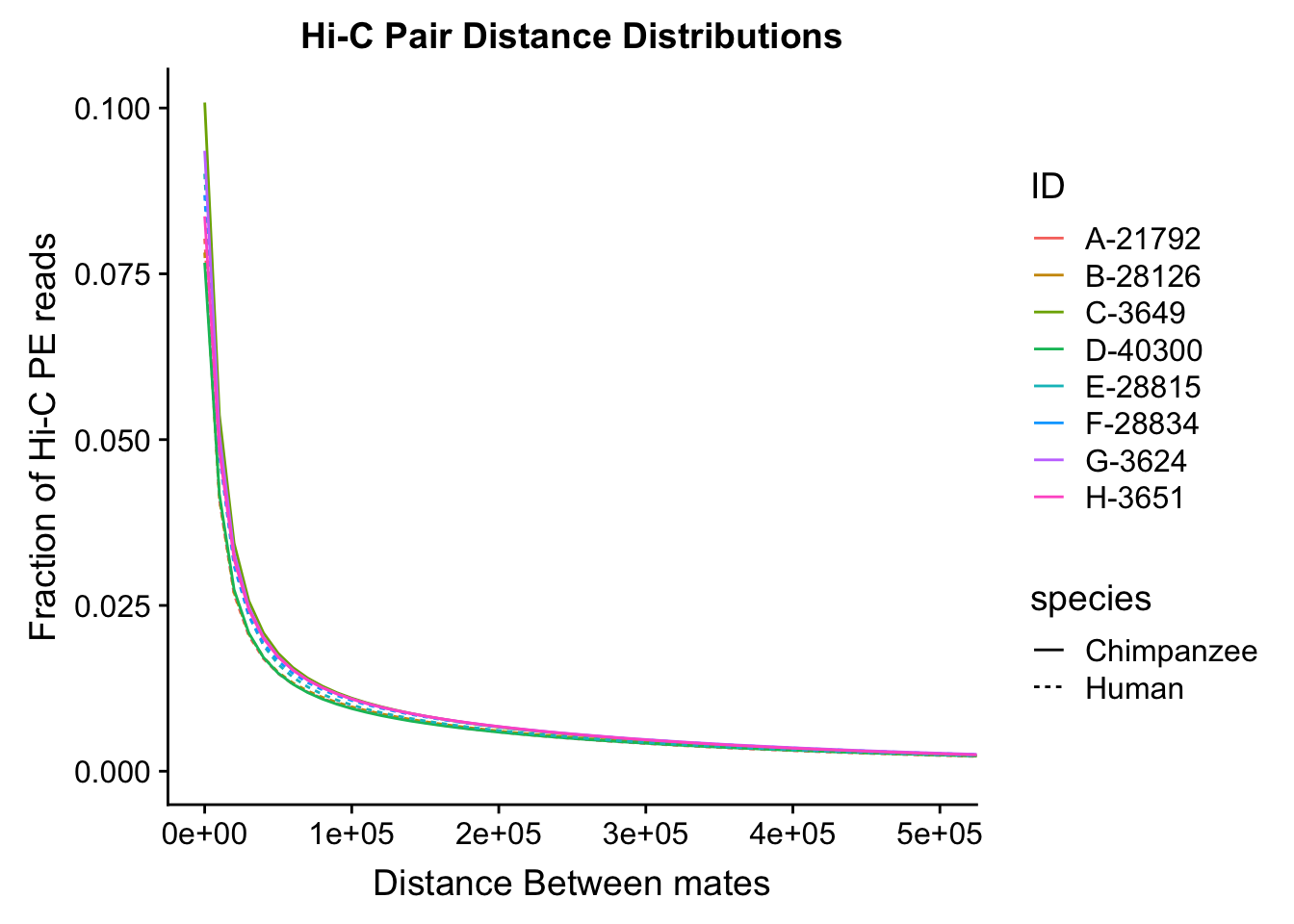
| Version | Author | Date |
|---|---|---|
| 3d877d7 | Ittai Eres | 2019-03-12 |
#Once again, there appears to be no clear separation between species here. There doesn't even appear to be a batch effect!
sessionInfo()R version 3.4.0 (2017-04-21)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] compiler stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] forcats_0.4.0 purrr_0.3.2 readr_1.3.1
[4] tibble_2.1.3 tidyverse_1.2.1 edgeR_3.20.9
[7] RColorBrewer_1.1-2 heatmaply_0.16.0 viridis_0.5.1
[10] viridisLite_0.3.0 stringr_1.4.0 gplots_3.0.1.1
[13] Hmisc_4.2-0 Formula_1.2-3 survival_2.44-1.1
[16] lattice_0.20-38 dplyr_0.8.3 plotly_4.9.0
[19] cowplot_0.9.4 ggplot2_3.2.1 reshape2_1.4.3
[22] data.table_1.12.0 tidyr_1.0.0 plyr_1.8.4
[25] limma_3.34.9
loaded via a namespace (and not attached):
[1] nlme_3.1-137 bitops_1.0-6 fs_1.3.1
[4] lubridate_1.7.4 webshot_0.5.1 httr_1.4.1
[7] rprojroot_1.3-2 tools_3.4.0 backports_1.1.4
[10] R6_2.4.0 rpart_4.1-15 KernSmooth_2.23-15
[13] lazyeval_0.2.2 colorspace_1.4-1 nnet_7.3-12
[16] withr_2.1.2 processx_3.4.1 tidyselect_0.2.5
[19] gridExtra_2.3 git2r_0.26.1 cli_1.1.0
[22] rvest_0.3.4 htmlTable_1.13.2 TSP_1.1-7
[25] xml2_1.2.2 labeling_0.3 caTools_1.17.1.2
[28] scales_1.0.0 checkmate_1.9.4 digest_0.6.18
[31] foreign_0.8-72 rmarkdown_1.12 base64enc_0.1-3
[34] pkgconfig_2.0.3 htmltools_0.3.6 highr_0.8
[37] readxl_1.3.1 htmlwidgets_1.3 rlang_0.4.0
[40] rstudioapi_0.10 shiny_1.3.2 generics_0.0.2
[43] jsonlite_1.6 crosstalk_1.0.0 gtools_3.8.1
[46] acepack_1.4.1 dendextend_1.12.0 magrittr_1.5
[49] Matrix_1.2-17 Rcpp_1.0.1 munsell_0.5.0
[52] lifecycle_0.1.0 stringi_1.4.3 whisker_0.4
[55] yaml_2.2.0 MASS_7.3-51.4 grid_3.4.0
[58] promises_1.0.1 gdata_2.18.0 crayon_1.3.4
[61] haven_2.1.1 splines_3.4.0 hms_0.5.1
[64] locfit_1.5-9.1 ps_1.3.0 zeallot_0.1.0
[67] knitr_1.22 pillar_1.4.2 codetools_0.2-16
[70] glue_1.3.1 gclus_1.3.2 evaluate_0.13
[73] latticeExtra_0.6-28 modelr_0.1.5 httpuv_1.5.2
[76] vctrs_0.2.0 foreach_1.4.7 cellranger_1.1.0
[79] gtable_0.3.0 assertthat_0.2.1 xfun_0.5
[82] mime_0.7 xtable_1.8-4 broom_0.5.2
[85] later_0.8.0 seriation_1.2-3 iterators_1.0.12
[88] registry_0.5-1 workflowr_1.4.0 cluster_2.0.7-1